Коллоквиум. коллок 2. Цепной механизм химических превращений
 Скачать 5.07 Mb. Скачать 5.07 Mb.
|

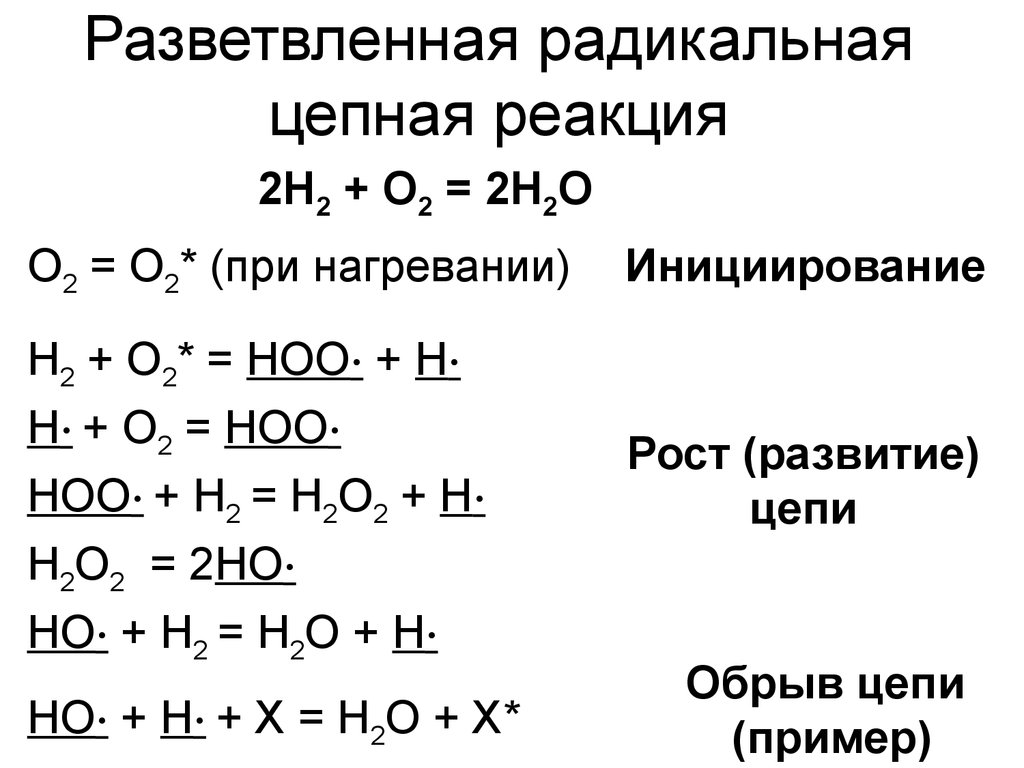
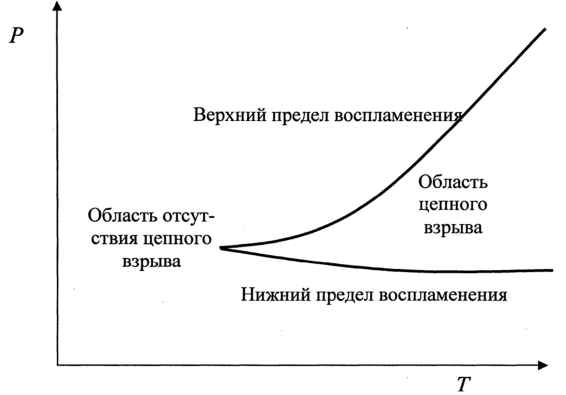
0 означает приблизительно постоянную концентрацию [X] « const.Цепной механизм химических превращений. Элементарные стадии зарождения, продолжения и обрыва цепи. Длина цепи неразветвленной цепной реакции. Приближенные методы химической кинетики: метод квазистационарных концентраций, квазиравновесное приближение. Кинетика неразветвленных цепных реакций на примере реакции образования фосгена. Разветвленные цепные реакции. Кинетика разветвленных цепных реакций. Вырожденное разветвление. Предельные явления в разветвленных цепных реакциях на примере окисления гидрида кремния. Полуостров воспламенения. Применение метода квазистационарных концентраций для описания предельных явлений в окрестностях первого и второго пределов воспламенения. Следует обратить внимание на то, что условием применения метода является сравнительная малость концентраций, а не относительные величины констант скоростей. Это объясняется тем, что константы скоростей последовательных стадий в общем случае могут иметь разные размерности из-за разных кинетических порядков. Тогда не имеет смысла сравнивать их численные значения. С другой стороны, относительно низкая концентрация означает короткий индукционный период, благодаря чему метод стационарных концентраций оказывается применим почти на всем протяжении реакции. Справедливость метода стационарных концентраций не может быть доказана для произвольной последовательности элементарных стадий, потому что невозможно решить произвольную систему дифференциальных уравнений. Однако опыт применения этого метода свидетельствует о его достаточной точности во всех реакциях с относительно низкой концентрацией промежуточных веществ. Рассмотрим применение метода к реакции А + В = Z с механизмом: 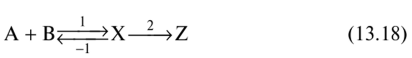 Для него есть той дифференциальных 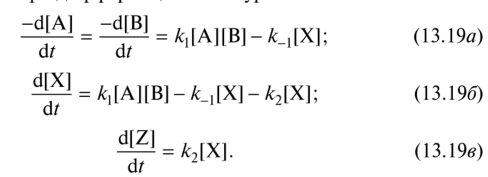 уравнения: уравнения:Чтобы выразить скорость образования Z через концентрации исходных веществ точной аналитической формулой, пришлось бы выразить концентрацию [X] в явном виде из уравнений (13.19я, б) и подставить ее в дифференциальное уравнение (13.19б) принимает вид: Отсюда: 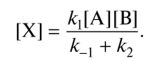 Подставив это выражение в (13.19в), получим: 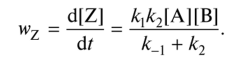 Сумма стадий (13.18) составляет реакцию А+В = Z. Если концентрация X много меньше концентраций А и В в любой момент времени реакции, то для этого стехиометрического уравнения верно: Поэтому можно записать для механизма (13.18): 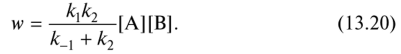 Это является теоретическим уравнением скорости рассматриваемой реакции — теоретическим в том смысле, что оно выведено из предполагаемого механизма. Предположим, что реакция (13.18) исследуется экспериментально путем слежения за концентрациями исходных веществ А и В и продукта Ъ и что неизвестно о существовании промежуточных частиц X. Из уравнения (13.20) ясно, что результатом исследования было бы эмпирическое уравнение скорости = = &[А][В], в котором коэффициент к в действительности имеет следующее выражение:  Пример 13.1. Для реакции разложения оксида азота (V): 2М205 = = 4Ы02 + 02 известно эмпирическое уравнение скорости = &[М205]. Считается, что она происходит по следующему механизму: 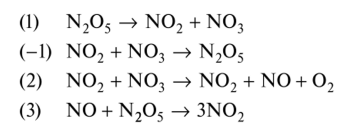 Методом квазистационарных концентраций вывести теоретическое уравнение скорости и выразить коэффициент скорости к через константы скоростей элементарных стадий. Решение. Заметим сначала, что стадия (-1) является обратной по отношению к стадии 1. Вместе они составляют двустороннюю реакцию. Заметим также, что имеются два сорта промежуточных частиц: N03 и N0. Частица 1М03 образуется в стадии 1 и расходуется в стадиях (-1) и 2. Напишем для них суммарное уравнение скорости образования и примем эту скорость равной нулю (по методу стационарных концентраций): 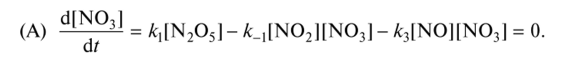 Вещество N0 образуется в стадии (2) и расходуется в стадии (3). Напишем для него суммарное уравнение скорости образования и также примем скорость равной нулю: 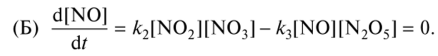 Исходное вещество ^05 расходуется в стадиях (1) и (3) и образуется в стадии (-1). Напишем для него суммарное уравнение скорости расходования: 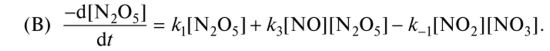 Выразим &3[М0]^205] из (Б): 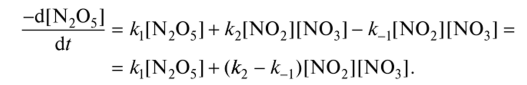 Выразим произведение [N02][N03] из (А): 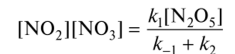 и подставим в предыдущее уравнение для скорости расходования -б[1Ч205]/с1/: 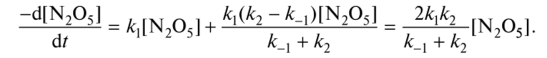 372 Это является теоретическим уравнением скорости расходования N205. Уравнение скорости реакции 214 205 = 414 02 + 02 получается делением на стехиометрический коэффициент 2: 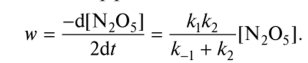 Сравнив его с эмпирическим уравнением скорости н> = /г[14205], находим к = кхк2/(к_х + к2). Фотохимические процессы. Называются реакции, которые протекают при воздействии облучения.  Основные законы фотохимии.  Первичные фотохимические процессы. Фотохимические реакции подразделяются на первичные и вторичные процессы, происходящие соответственно на световых или темновых стадиях. Первичные процессы связаны с поглощением молекулами кванта световой энергии - фотона, что может привести к образованию реакционноспособных промежуточных соединений, которые затем участвуют во вторичных процессах, имеющих термическую природу и не требующих воздействия света или другого излучения Проиллюстрировать первичные и вторичные процессы можно на примере разложения иодида водорода. В отсутствие света происходит термическая реакция 2HJ ® H2 + J2 Если же на систему действует свет подходящей длины волны, то идут следующие процессы: HJ + hn ® H + J фотохимическая реакция, первичный процесс H + HJ ® H2 + J ü термические реакции; J + J ® J2 þ вторичные процессы Хотя конечные продукты превращений в химическом и в фотохимическом процессах совпадают, механизмы этих процессов различны. Квантовый выход.   Кинетика и определение первичного квантового выхода фотохимических реакций. Рассмотрим возможные пути развития фотохимической реакции после активации реагирующей частицы. Далеко не всегда следующим за активацией актом является процесс, в результате которого возникают продукты. Такие процессы называют первичными. К первичным процессам относятся: 1) флюоресценция: А" —> А + h v; 2) дезактивация при соударении со стенкой сосуда или другой неактивной молекулой: А' + />тшшя) А + /(стетка); 3) прямая диссоциация А' —> D, -г D2 ; 4) изомеризация с образованием другой конформации: А* —> В; 5) реакция с другой молекулой: А' + В —» С. Если доминирующим из вышеприведенных будет процесс 5, то квантовый выход будет близок к 1, однако эксперименты показывают, что лишь единичные реакции идут таким образом. Чаще всего преобладает процесс диссоциации 3. При таком механизме реакции можно ввести понятие первичного квантового выхода: Первичный квантовый выход, таким образом, может быть либо равен 1, если процессы 1 и 2 нс проходят, либо < 1, при их наличии. Представление о кинетике реакций в твердых телах. Одна из первых формул, описывающих кинетику твердофазовых реакций, было предложена В. Яндером. Уравнение Яндера, выведенное исходя из закона Фика, для стадии твердофазовой реакции, контролируемой процессом диффузии, имеет для изотермических условий вид: Где х — толщина слоя продукта реакции на покрываемом, менее подвижном компоненте; τ — время; D — коэффициент диффузии при данной температуре; с0 — концентрация диффундирующего компонента на границе со слоем продукта реакции. Принимая величины D и с0 постоянными и обозначая Dc=k после интегрирования уравнения (1) получаем: x2 = 2kτ (2) т.е. в изотермических условиях квадрат толщины слоя продукта реакции пропорционален времени. Поскольку измерить толщину слоя продукта реакции практически очень трудно, особенно в порошкообразных смесях, Яндер впоследствии видоизменил зависимость (2) и выразил ее через степень химического превращения компонента, покрываемого слоем продукта реакции, что достаточно легко определить обычными методами количественного анализа. Видоизмененная формула Яндера для изотермических условий имеет вид: 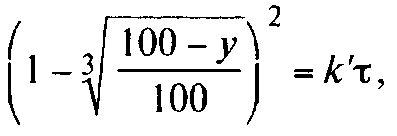 (3) (3)Где y -степень превращения покрываемого компонента, т.е. его количество, вошедшее в реакцию, мас.%; k -постоянная. Применимость уравнения (3) для описания конкретного твердофазового процесса проверяется с помощью графического построения зависимости величины (l - 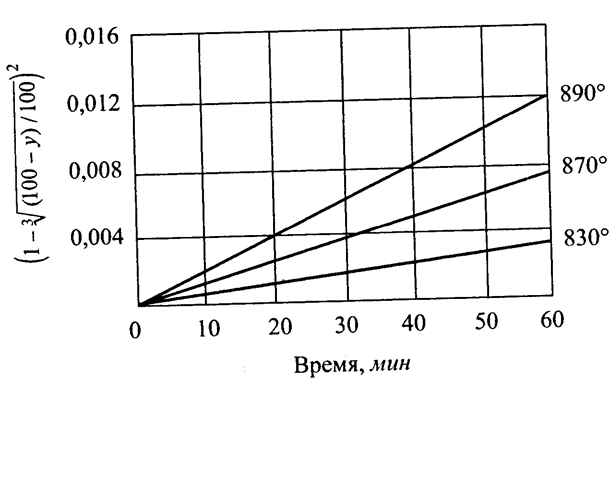 Рис 64 Зависимость параметров (l - A.M. Гинстлинг и Б.И. Броунштейн рассмотрели кинетическую модель процесса твердофазового взаимодействия, учитывающую сферичность реагирующих зерен в смеси, и предложили следующее уравнение кинетики твердофазовых реакций для изотермического процесса: 1- 2/3G-(1-G)2/3 = k''τ, (4) Где G — степень превращения покрываемого компонента в долях единицы; τ — время; k постоянная. Уравнение (4) оказалось более строгим и для многих систем дает более точные результаты, особенно при больших степенях превращения компонентов и больших значениях τ, чем уравнения Яндера (рис.65). Следует отметить, что уравнения Яндера и Гинстлинга — Броунштейна, основанные на одном и том же исходном положении: скорость твердофазовой реакции обратно пропорциональна толщине слоя продукта реакции, были получены исходя из предположения об образовании продукта реакции путем односторонней диффузии покрывающего компонента в глубь зерен покрываемого компонента. 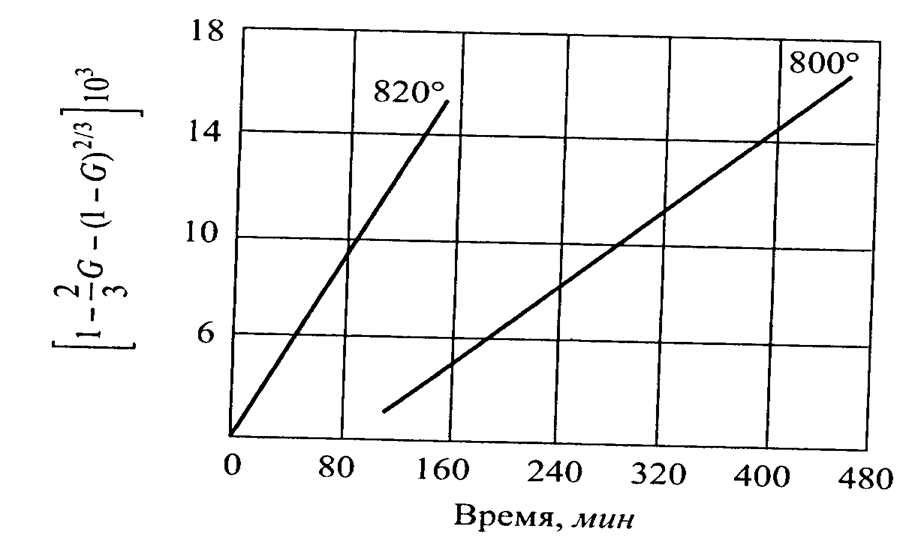 Рис 65 Зависимость параметров (l - Поскольку в реальных условиях твердофазовые реакции могут осуществляться также за счет противодиффузии реагентов через слой продукта реакции, рассмотренные выше кинетические уравнения, не являются универсальными и удовлетворительно описывают кинетику отдельных твердофазовых реакций или их стадий. Факторы, влияющие на скорость твердофазовых реакций Температура. Температура - один из важнейших факторов, влияющих на скорость твердофазовых реакций. Если процесс протекает в диффузионной области (а это справедливо для большинства реакций в силикатных системах), то скорость реакции определяется температурной зависимостью коэффициента диффузии:  Где А - коэффициент, формально равный коэффициенту диффузии при Т=∞ Q- энергия активации процесса диффузии или энергия «разрыхления» решетки, зависящая от сил связи между ее структурными элементами; R — газовая постоянная; Т — абсолютная температура. Гранулометрический состав порошков. Твердофазовые реакции обычно протекают в порошкообразных смесях, в которых отдельные зерна соприкасаются между собой лишь небольшой частью поверхности. Так, в реальных порошкообразных массах поверхность контакта между зернами составляет от 10-4 до 10-7 (в долях от общей поверхности). Поскольку чисто твердофазовые реакции (без участия жидкой и газообразной фаз) протекают именно на поверхности контакта зерен, то величина площади их соприкосновения очень сильно влияет на скорость подобных реакций. При прочих равных условиях скорость чисто твердофазового взаимодействия пропорциональна поверхности контакта реагирующих веществ и увеличение поверхности контакта между зернами способствует ускорению твердофазовых реакций. Давление. Важным фактором, влияющим на скорость твердофазовых реакций, является давление, прикладываемое к порошкообразным смесям, поскольку площадь поверхности контакта между зернами пропорциональна давлению сжатия. Увеличение поверхности контакта порошков при сжатии происходит за счет возрастания площади имеющихся контактных участков в результате пластической иди хрупкой деформации, а также за счет возникновения новых мест контактов. Присутствие газовой или жидкой фазы. Большое влияние на скорость твердофазовых реакций оказывает присутствие в системе реагирующих веществ газовой или жидкой фазы, которые сами непосредственно в реакции участия не принимают. Катализ. Лекция Общие представления о механизме и кинетике каталитических реакций. При этом все многообразие механизмов каталитических реакций может быть сведено к двум схемам. Согласно первой из них, катализатор (К) является переносчиком атомов, атомных групп или электронов от молекулы одного реагента (А) к молекуле другого (В). Так, механизм реакции А + В = С + D может быть представлен совокупностью стадий: 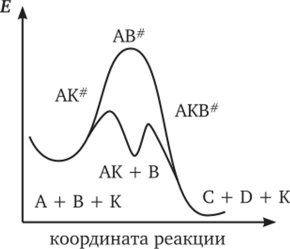 По этой причине подобный механизм каталитического действия называют стадийным. На первой стадии механизма образуется промежуточное соединение (АК) и один из продуктов реакции (С). На второй стадии при взаимодействии промежуточного соединения (АХ) с молекулой второго реагента (В) получается второй продукт реакции (D), а активный центр регенерируется и вступает в новый каталитический цикл. Примером такого механизма является реакция окисления S02 кислородом воздуха на поверхности гетерогенного катализатора (в промышленности это оксид молибдена, нанесенный на поверхность пористого оксида алюминия): 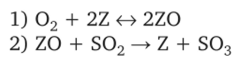 Здесь Z — активный центр на поверхности катализатора. Первая стадия механизма — хемосорбция молекулы кислорода, сопровождающаяся диссоциацией на атомы. Во второй стадии происходят образование продукта реакции S03 и регенерация активного центра. На рис. 6.15 приведена энергетическая диаграмма реакции. Видно, что в присутствии катализатора процесс идет с меньшей энергией активации (энергией Гиббса активации), чем без него. 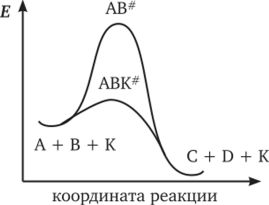 Рис. 6.16. Энергетическая диаграмма слитной схемы механизма (знаком # обозначены активированные комплексы) Суть второй схемы каталитического действия заключается в обратимом образовании промежуточного соединения из активного центра катализатора и молекул обоих реагентов: А и В. Далее это соединение распадается на продукты и «свободный» активный центр катализатора, последний вновь вступает в цикл превращений и т. д. Механизм такого каталитического действия называют слитным. Последовательность стадий реакции в этом случае представляют следующей схемой: 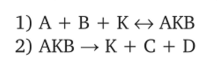 В качестве примера можно привести реакцию жидкофазного гидрирования олефинов на поверхности гетерогенных катализаторов — металлов платиновой группы: 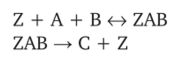 Здесь Z — активный центр (атом) платинового металла, ZAB — л-комплекс из атома металла, молекулы олефина (А) и молекулы водорода (В). Обе приведенные выше схемы отражают общую и характерную особенность механизма каталитических реакций, а именно — его цикличность. Цикличность в катализе отличается от цикличности в цепных реакциях. Цепные реакции представляют собой последовательность стадий, в каждой из которых участвует вновь образующаяся активная частица — радикал. В каталитических реакциях один и тот же активный центр или молекула катализатора может многократно (103—1011 раз) вступать в химическое взаимодействие с молекулами реагентов. Следует отметить, что кинетические закономерности каталитических реакций не исчерпываются приведенными выше двумя схемами. Многие каталитические процессы включают в себя десятки параллельно-последовательных стадий. Установить механизм таких процессов возможно лишь на основании детальных кинетических исследований и в сочетании с другими физико-химическими методами исследования. Последние (спектральные, электрохимические, термохимические и др.) методы позволяют установить наличие и химическое строение промежуточных соединений. Как уже отмечалось, энергия активации каталитической реакции всегда ниже, чем реакции в отсутствии катализатора. Роль катализа в современной химической технологии. Роль катализа в современной технологии трудно переоценить. Катализ — это изменение скорости химических реакций под влиянием особых веществ — катализаторов. Катализатор, помогая осуществить химическую реакцию, по окончании ее выделяется в неизменном виде; таким образом, роль катализатора сводится к изменению пути протекания химических реакций. На основе катализа созданы перспективные способы производства моторных топлив из угля, сланцев и торфа; широкое применение находят каталитические процессы гидрирования жиров в пищевой промышленности. Все больше используется катализ для охраны окружающей среды от загрязнений. сточными водами, вредными промышленными автомобильными газами. Гомогенный катализ. Гомогенный катализ, ускорение химической реакции в присутствии катализатора, который находится в одной фазе с исходными реагентами (субстратами) в газовой фазе или растворе. При гомогенном катализе, как и при гетерогенном катализе, катализатор в реакции не расходуется, однако является ее необходимым участником; без катализатора реакция протекает гораздо медленнее или не идет вовсе. Общие кинетические закономерности. Химическая кинетика рассматривает и устанавливает зависимости скорости химических реакций от концентраций реагентов, температуры и других внешних условий. Закономерности всех процессов могут быть сформулированы в виде общего закона: скорость (интенсивность) процесса прямо пропорциональна движущей силе и обратно пропорциональна сопротивлению: Гомогенно-каталитическое разложение пероксида водорода.  Кислотно-основной катализ.  Кинетика и механизм реакций общего кислотного и специфического кислотного катализа.   Уравнение Бренстеда, его использование в химической кинетике. Уравнение Бренстеда — уравнение, количественно описывающее зависимость скорости катализируемых кислотами или основаниями реакций от природы катализатора. Впервые установлено в 1924 году Брёнстедом и Педерсеном. Для кислотного катализа уравнение выражается в виде: где {\displaystyle k_{a}}ka — каталитическая константа скорости реакции, {\displaystyle K_{a}}Ka — константа кислотности кислоты, выступающей в качестве катализатора, {\displaystyle k_{0}}k0 — константа скорости некатализируемой реакции, {\displaystyle \alpha }a — константа, характеризующая реакционную серию и отражающая чувствительность скорости реакции к смене катализатора. В общем виде уравнение записывается в виде: Для основного катализа уравнение выражается в аналогичном виде:{\displaystyle \log(k)=\alpha \cdot \log(K_{a})+C}  Кинетика и механизм ферментативных реакций. Кинетика ферментативных реакций – наука о скоростях ферментативных реакций, их зависимости от различных факторов. Скорость ферментативной реакции определяется химическим количеством прореагировавшего субстрата или образовавшегося продукта реакции в единицу времени в единице объема при определенных условиях: где v – скорость ферментативной реакции, Скорость ферментативной реакции зависит от природы фермента, которая определяет его активность. Чем выше активность фермента, тем выше скорость реакции. Активность фермента определяют по скорости реакции, катализируемой ферментом. Мерой активности фермента является одна стандартная единица активности фермента. Одна стандартная единица активности фермента – это такое количество фермента, которое катализирует превращение 1 мкмоль субстрата за 1 минуту. В процессе ферментативной реакции фермент (Е) взаимодействует с субстратом (S), в результате образуется фермент-субстратный комплекс, который затем распадается с высвобождением фермента и продукта (Р) реакции: Скорость ферментативной реакции зависит от многих факторов: от концентрации субстрата и фермента, температуры, рН среды, наличия различных регуляторных веществ,способных увеличивать или снижать активность ферментов. 1 )Зависимость скорости ферментативной реакции от концентрации фермента 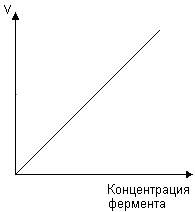 2)Зависимость скорости ферментативной реакции от температуры 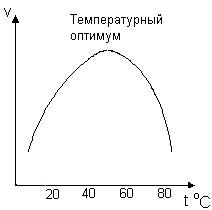 При низких температурах (приблизительно до 40 – 50 оС) повышение температуры на каждые 10 оС в соответствии с правилом Вант-Гоффа сопровождается увеличением скорости химической реакции в 2 – 4 раза. 3) График зависимости ферментативной активности от рН. 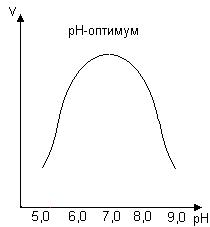 Уравнение Михаэлиса – Ментен.  Ингибированные ферментативные реакции. Вещества, присутствие которых в системе понижает активность фермента, т. е. уменьшает скорость реакции, называются ингибиторами. 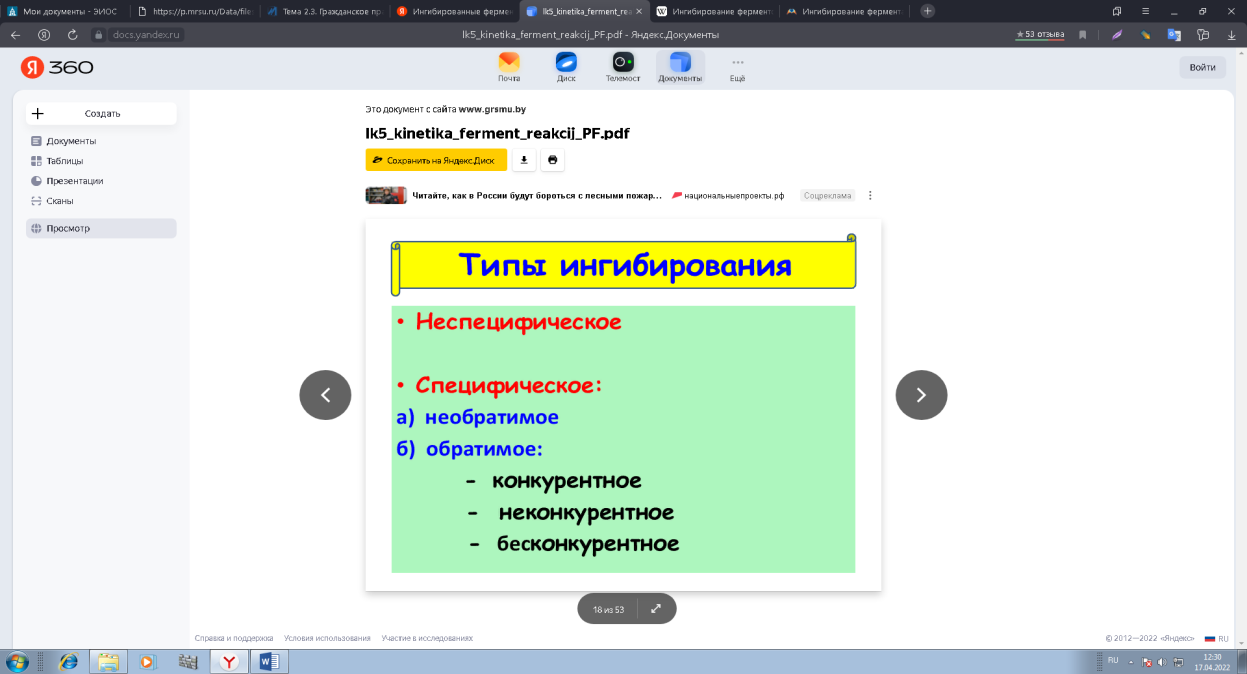 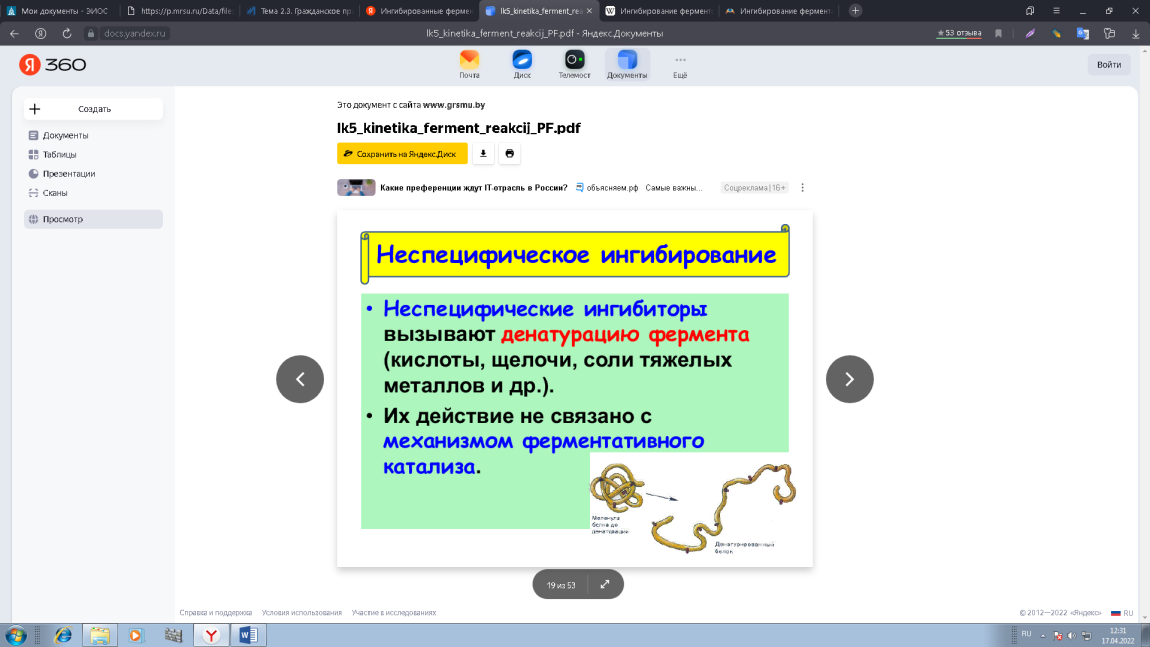 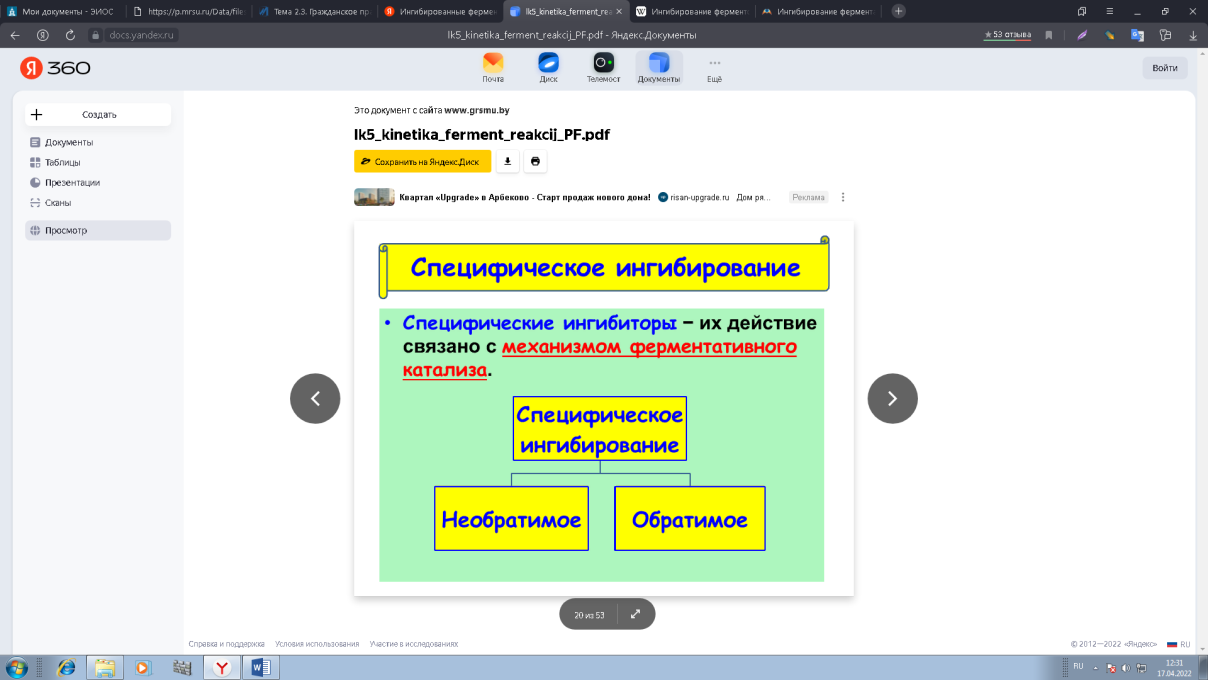 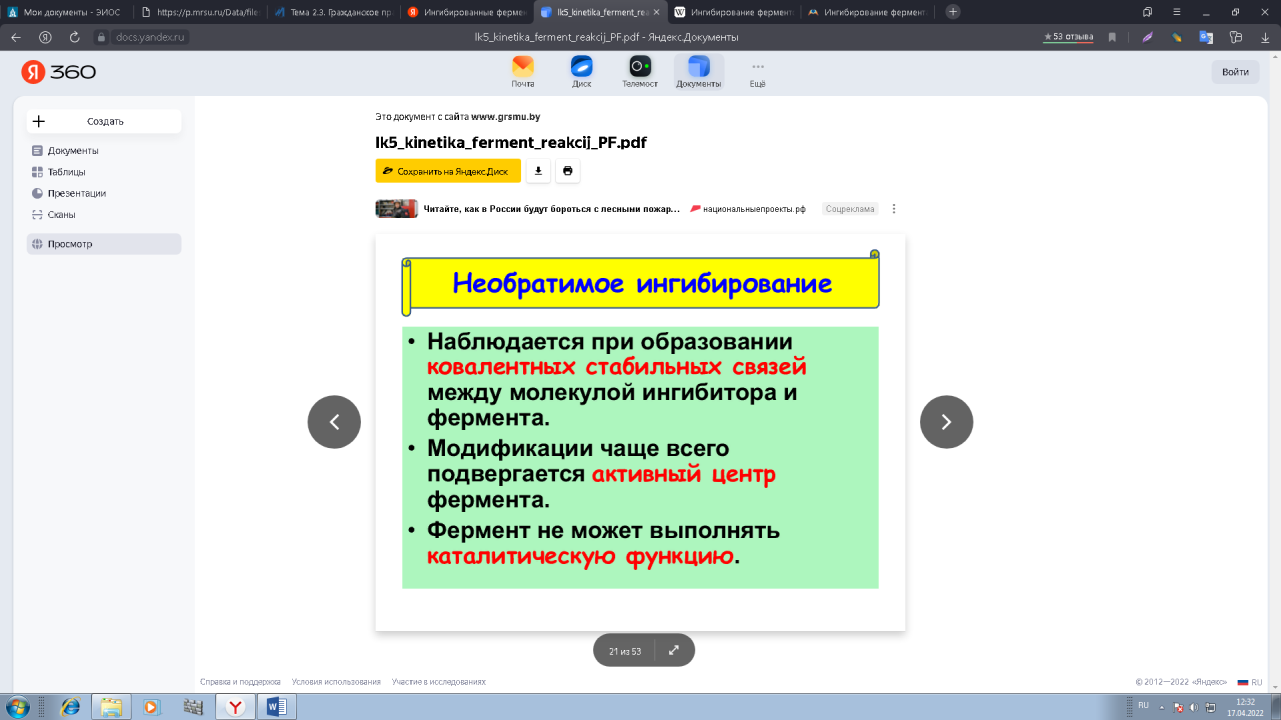 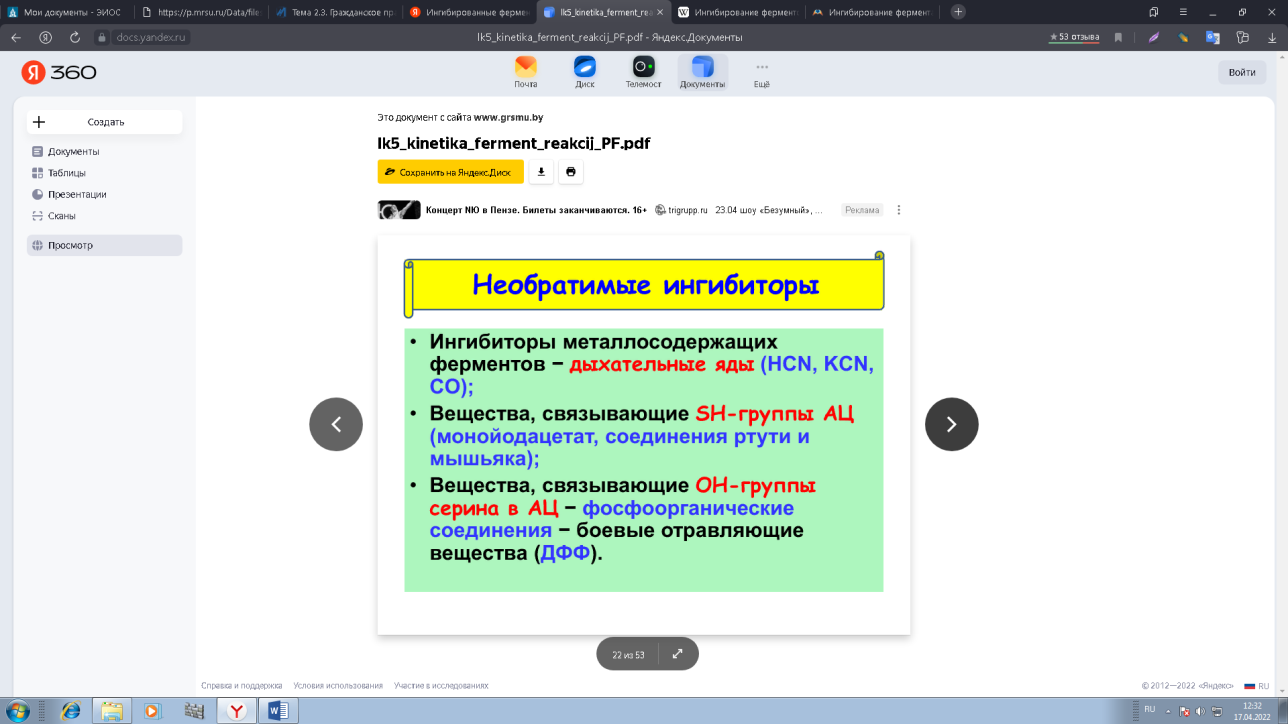 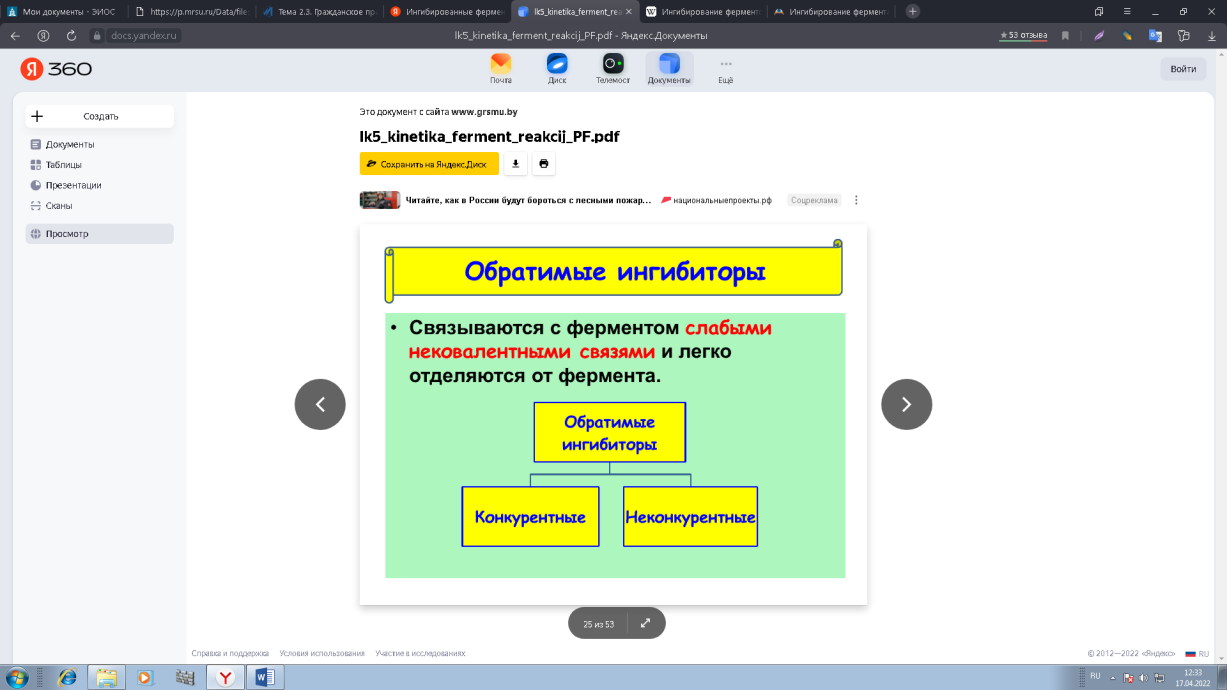 Конкурентный ингибитор – это соединение, обладающее структурным сходством с субстратом. • ингибитор способен взаимодействовать с активным центром фермента, конкурируя с истинным субстратом • Пример конкурентного ингибирования – ингибирование сукцинатдегдрогеназы малоновой кислотой Неконкурентный ингибитор не является структурным аналогом субстрата; • Ингибитор присоединяется не к активному центру, а к другому участку молекулы фермента; Бесконкурентный тип ингибирования наблюдается в случае, когда ингибитор способен связываться исключительно с фермент- субстратным комплексом Гетерогенный катализ. Гетерогенный катализ – каталитические реакции, идущие на поверхности раздела фаз, образуемых катализатором и реагирующими веществами. Механизм гетерогенно-каталитических процессов значительно более сложен, чем в случае гомогенного катализа. В каждой гетерогенно-каталитической реакции можно выделить как минимум шесть стадий: 1. Диффузия исходных веществ к поверхности катализатора. 2. Адсорбция исходных веществ на поверхности с образованием некоторого промежуточного соединения: А + В + К ––> АВК 3. Активация адсорбированного состояния (необходимая для этого энергия есть истинная энергия активации процесса): АВК ––> АВК# 4. Распад активированного комплекса с образованием адсорбированных продуктов реакции: АВК# ––> СDК 5. Десорбция продуктов реакции с поверхности катализатора. СDК ––> С + D + К 6. Диффузия продуктов реакции от поверхности катализатора. Специфической особенностью гетерокаталитических процессов является способность катализатора к промотированию и отравлению. Энергия активации гетерогенных каталитических реакций. В большинстве случаев для константы скорости гетерогенной каталитической реакции выполняется уравнение Аррениуса: из которого обычным путем можно определить Еа. Для одинаковых процессов Еа, полученная по уравнению Аррениуса гетерогенной реакции ниже, чем Еа гомогенной некаталитической реакции. Ускоряющее действие гетерогенного катализатора обусловлено экзотермическим процессом адсорбции на поверхности, вследствие чего снижается Еа реакции. Роль диффузии в кинетике гетерогенных реакций. Результирующая скорость гетерогенной реакции определяется самой медленной стадией. Чаще всего такой стадией является именно диффузия, поэтому она играет очень важную роль в кинетике гетерогенных процессов. Диффузия — это самопроизвольное перемещение частиц {молекул) из области с более высокой в область с более низкой концентрацией. В основе ее — хаотичное тепловое движение данных частиц. Режимы протекания реакций, кинетическая область, область внутренней и внешней диффузии.  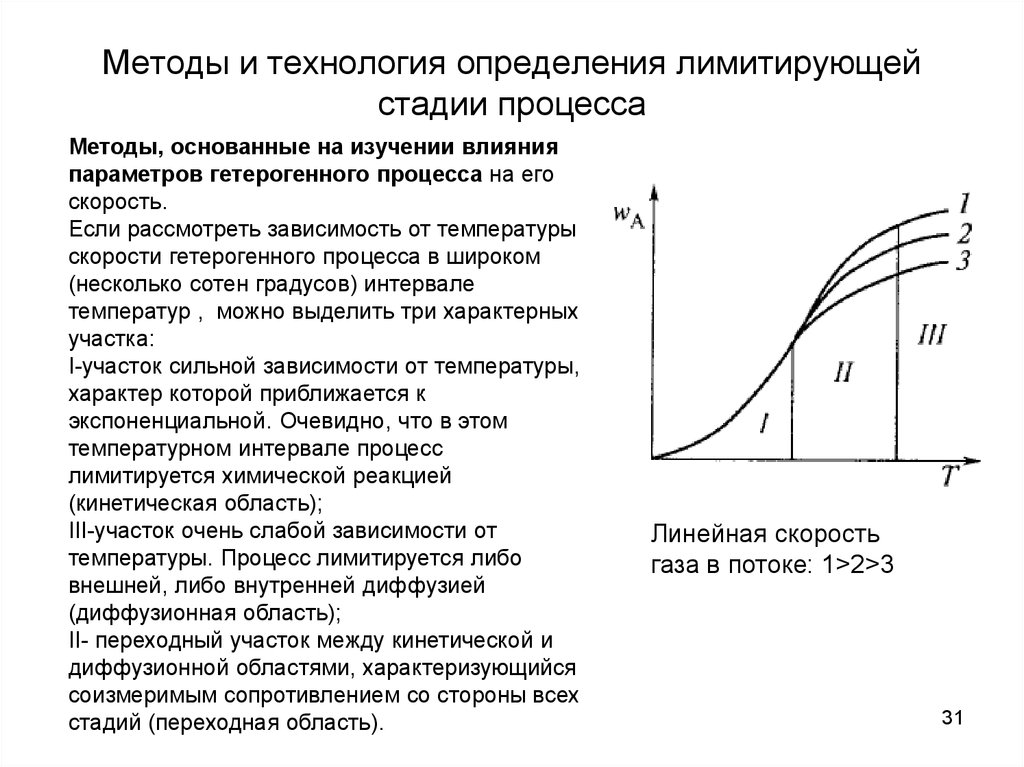 Влияние радиуса пор и размера гранул катализатора на режимы протеканий реакций. Кинетика гетерогенных каталитических реакций в статических условиях. 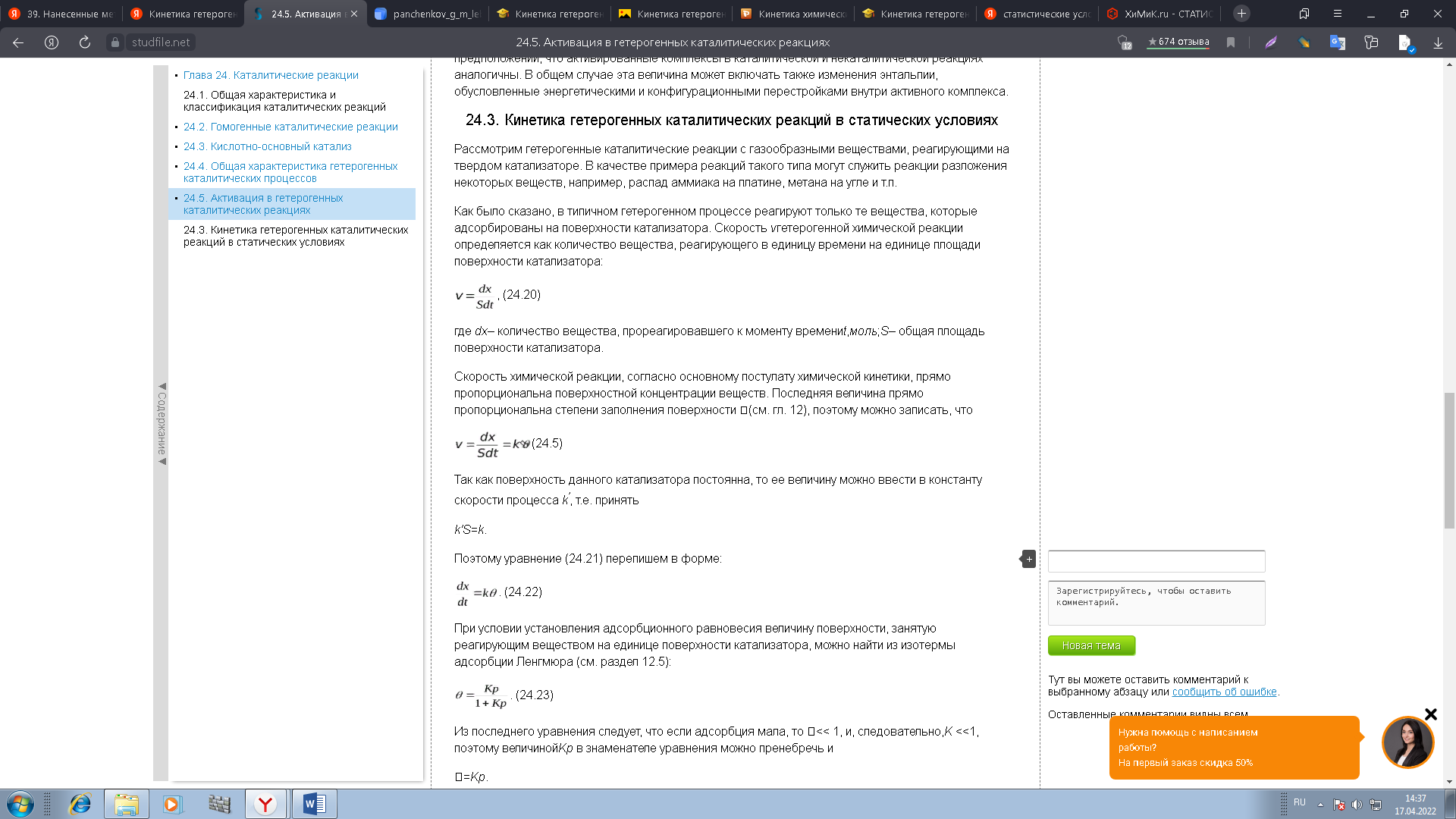 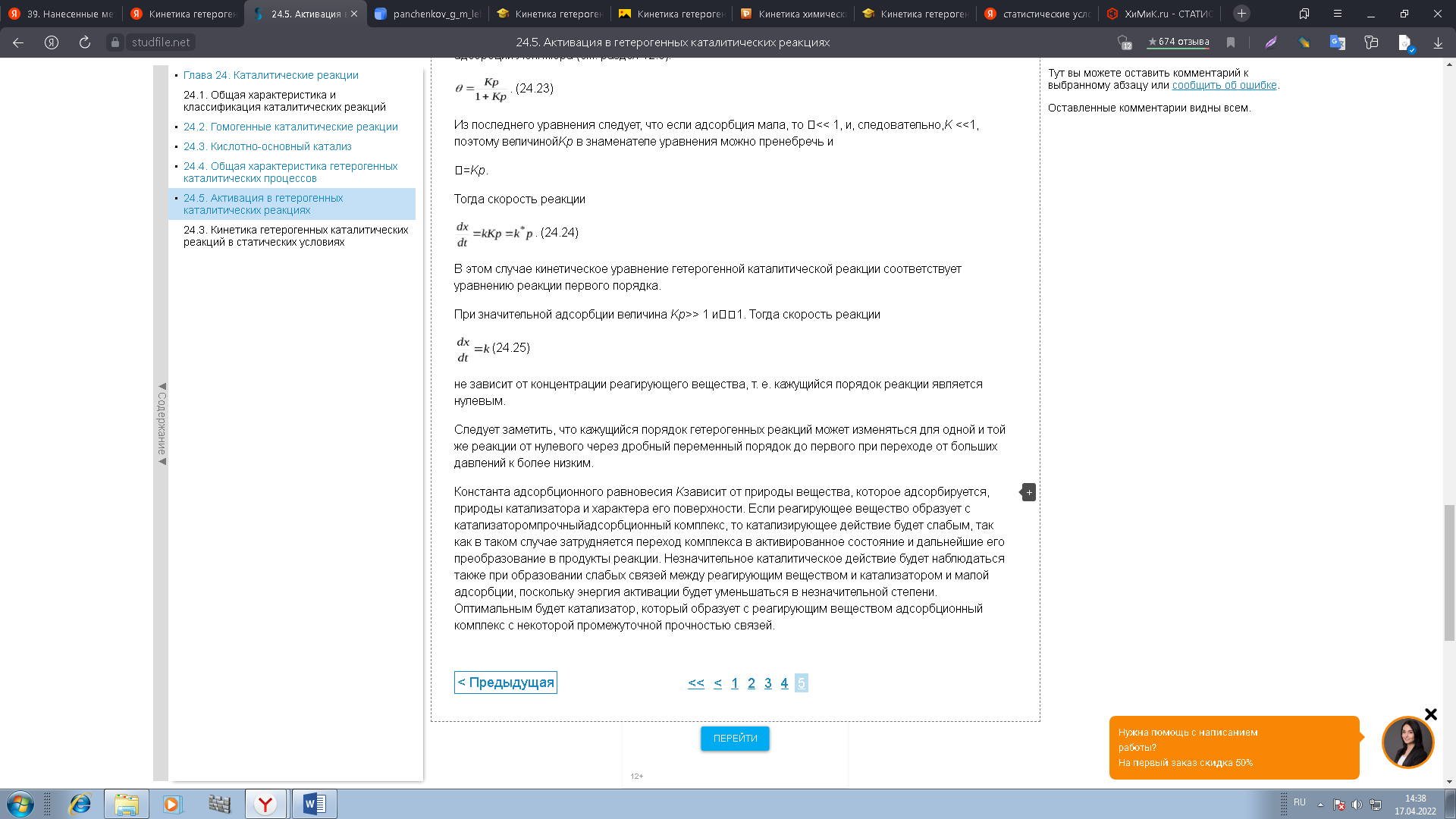 Теория мультиплетов Баландина. Сопоставление структуры и состава активного центра со строением реагирующих молекул рассматривает мультиплетная теория. А. А. Баландин в 1929 г. сформулировал основные положения мультиплетной теории: 1) Первым исходным положением мультиплетной теории являлся принцип геометрического соответствия: активный центр катализатора представляет собой совокупность определенного числа адсорбционных центров, расположенных на поверхности в геометрическом соответствии со строением реагирующей молекулы. 2) При адсорбции реагирующих молекул на активном центре образуется мультиплетный комплекс, в результате чего происходит перераспределение связей, приводящее к образованию продуктов реакции. Область применения теории мультиплетов. (??)Мультиплетная теория применяется при сопоставлении структуры и состава активного центра со строением реагирующих молекул. Первая группа экспериментальных фактов свидетельствует, что строение молекулы может в значительной степени определять характер каталитического превращения. Пример: дегидрироваться способны только углеводороды с шестичленными циклами, способные давать ароматическое кольцо. 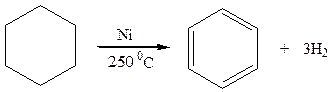 циклогексан бензол 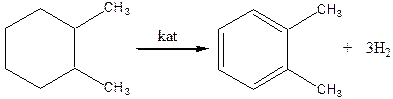 1,2-диметилциклогексан о-ксилол 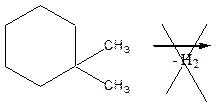 1,1-диметилциклогексан Вторая группа фактов свидетельствует, что строение самого катализатора влияет на процесс катализа. Пример: катализаторами дегидрирования являются металлы с определенным кристаллографическим строением и межатомными расстояниями в решетке Химическая теория катализа.     Нанесенные металлические катализаторы. НАНЕСEННЫЕ КАТАЛИЗАТОРЫ, содержат активный компонент, нанесенный на дисперсное или пористое в-во-носитель. Использование нанесенных катализаторов позволяет увеличить поверхность работающего катализатора, экономит дорогостоящие в-ва (напр., Pt, Pd, Ag), предотвращает рекристаллизацию и спекание активного компонента при высоких температурах, удлиняет срок работы катализатора, а в ряде случаев стабилизирует его в определенной хим. форме. Носитель должен обладать необходимыми хим. св-вами и адгезией, позволяющими удерживать на своей пов-сти активный компонент, обеспечивать доступ реагирующего в-ва к активным центрам катализатора, быть термически и химически устойчивым в условиях катализа и регенерации нанесенных катализаторов. Кол-во активного компонента в нанесенных катализаторах обычно значительно меньше кол-ва носителя. В качестве носителей применяют искусственные (активные угли, силикагель, Аl2О3, алюмосиликат, MgO, ZrO2) и (реже) естественные (прир. глины, пемза, диатомит, асбест) твердые тела с высоко развитой уд. пов-стью и пористостью. Однако в случае многостадийных р-ций, когда целевой продукт может подвергаться дальнейшим нежелат. превращениям, высокая пористость нанесенных катализаторов оказывается невыгодной вследствие возможности перехода р-ции во внутри-диффузионную область (см. Катализ, Катализаторы). Наиб. распространенный способ получения нанесенных катализаторов-пропитка носителя р-ром, содержащим активные компоненты катализатора, с послед, сушкой и прокаливанием. Для получения оксидных нанесенных катализаторов обычно применяют соли, анионы к-рых разлагаются при нагр. (нитраты, карбонаты, формиаты и т.п.); для получения металлических необходимо восстановление катализатора, пропитанного ранее р-ром соли. Физические основы теории активных ансамблей. 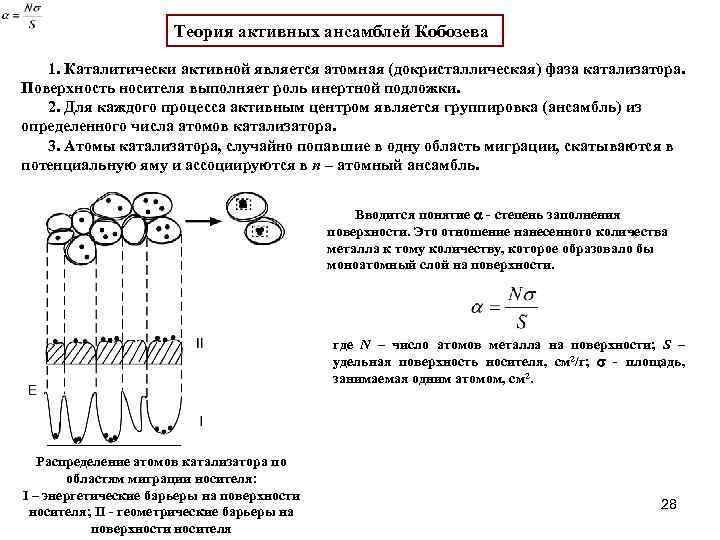 Общая и удельная активности катализатора (?????) продолжение 40 вопроса 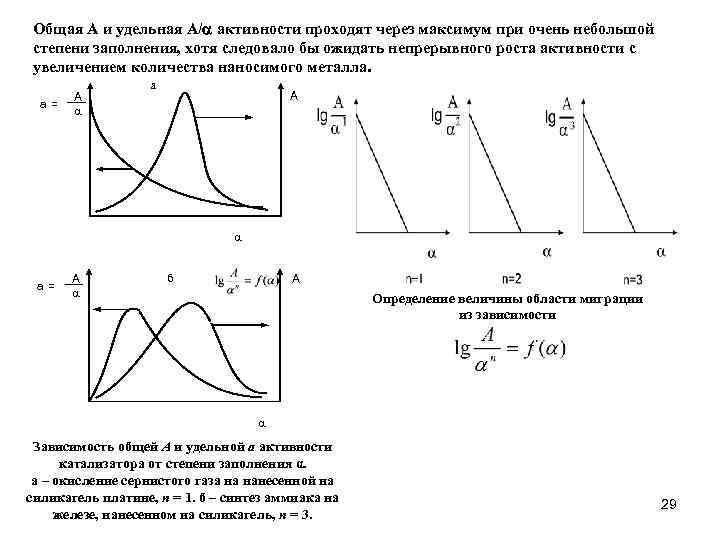 Применение гетерогенного катализа в промышленности. С помощью гетерогенного катализа в промышленности основного органического и нефтехимического синтеза осуществляются процессы гидрирования и дегидрирования, многие реакции окисления и окислительного аммонолиза, гидратации и дегидратации, алкилирования и т. д.  |