Хромосомные мутации. Хромосомные аберрации Механизмы хромосомных аберраций
 Скачать 299.51 Kb. Скачать 299.51 Kb.
|

|
Введение. Термин «мутации» используется для обозначения любых стабильных врожденных генетических изменений. В зависимости от того, в каких структурах наследственного аппарата гаметы произошла мутация возможно развитие различных мутаций: генных, хромосомных или геномных. Нормальные клетки человека имеют 46 хромосом: 22 пары аутосом и две половые хромосомы. По одной хромосоме в каждой паре человек получает от каждого родителя. Гаметы являются носителями генов, унаследованных ими не только от родителей, но и от всех отдаленных предков. Тяжелые повреждения гамет могут вести к их гибели, развитию бесплодия, спонтанных абортов и выкидышей. Гамета с дефектом гена или генов может стать источником наследственных пороков развития или заболеваний. Хромосомные аберрации Механизмы хромосомных аберраций Нерасхождение гомологичных хромосом происходит при первом мейотическом делении, в результате которого образуются гаметы. Таким образом, одна гамета получает пары хромосом, а вторая не получает ни одной. После второго мейотическоо деления образующиеся гаметы содержат 24 или 22 хромосомы. Такие гаметы называются анеуплоидными (количество хромосом в них не равно 23, нормальному гаплоидному набору человека). При оплодотворении анэуплоидной гаметы нормальной образуется анэуплоидная зигота, имеющая или три одинаковые хромосомы (трисомия) или только одну (моносомия). Трисомия или моносомия половых хромосом обычно совместима с жизнью, например, синдром Клайнфельтера (ХХY), синдром Тернера (ХО). При аутосомной моносомии теряется огромное количество генетического материала, поэтому она обычно летальна. Некоторые аутосомальные трисомии совместимы с жизнью, но связаны с тяжелыми нарушениями. Нерасхождение хромосом на ранних этапах развития зиготы приводит и развитию мозаицизма: присутствию в организме человека двух или более генетически различных клеточных популяций. Фенотипические проявления нарушений колеблятся от нормы к классическим проявлениям хромосомных аберраций. Так, например, у человека с мозаичным кариотипом Тернера (45, Х/46, ХХ) проявления будут колебаться от признаков нормальной женщины до проявлений классического синдрома Тернера (45, ХО). Делеция - это потеря части хромосомы в результате ее разрыва. Большинство делеций летальны в результате потери огромной части генетического материала. Делеция короткого плеча 4 и 5 хромосом приводит к развитию хорошо изученных синдромов (синдром Вольфа и синдром кошачьего крика соответственно). Частичная делеция хромосом часто определяется в опухолевых клетках (см. Общее учение об опухолях и Опухоли из кроветворной ткани). Транслокация - это перенос отдельного сегмента одной хромосомы в другую хромосому. При сбаласнсированной транслокации весь генетический материал сохраняется и остается фунционально способным, поэтому фенотипических проявлений нет. Наиболее известной сбалансированной транслокацией является перенос целой 21-ой хромосомы в 14-ую хромосому. Такие люди имеют 45 хромосом с отсутствием одной 14 и одной 21 хромосомы и наличием ненормальной большой хромосомы, содержащей материал обоих хромосом (14 и 21); в основном поражаются мужчины, кариотип которых обзначается как 45, XY, t (14; 21). У таких людей могут формироваться аномальные гаметы, в результате чего у потомков может развиваться моносомия 21 (несовместимая с жизнью) или транслокационный тип синдрома Дауна. Другие типы сбалансированных транслокаций являются причиной привычных, или повторяющихся абортов. Изменения в единичном нуклеотиде - проявляются нарушением расшифровки триплетного кода, но не вызывает определяемых структурных изменений в хромосомах. Эти нарушения названы болезнями единичного нуклеотида. Причины хромосомных аберраций Большинство хромосомных дефектов происходит «случайно», то есть без видимой причины, но в некоторых случаях она может быть установлена: Увеличение возраста матери - при увеличении возраста матери происходит учащение случаев не расхождения хромосом, подтверждением чего является трисомия 21-ой хромосомы (синдром Дауна). Риск трисомии 21-ой хромосомы, который составляет 1:1500 живорожденных у женщин в возрасте моложе 30 лет, увеличивается до 1:30 у женщин старше 45 лет. Поэтому беременным женщинам старше 35 лет настоятельно рекомендуется производить хромосомный анализ клеток плода, полученных при амниоцентезе. Увеличение материнского возраста также связано с другими синдромами не расхождения хромосом, например, синдром Кляйнфельтера. Ионизирующее излучение - увеличение количества случаев хромосомных аберраций наблюдалось у людей, выживших после атомной бомбардировки Хиросимы и Нагасаки. Не существует «безопасной» дозы ионизирующей радиации, поэтому диагностическое рентген-исследование брюшной полости у беременных должно производиться только в исключительных случаях. Медикаменты и другие химические вещества являются менее частой причиной структурных изменений хромосом. Использование противоопухолевых лекарств, которые нарушают синтез ДНК, в ранние сроки беременности могут привести к гибели плода. Множество широко используемых лекарств, в том числе и аспирин, вызывают изменения кариотипа в тканевых культурах. Наиболее частые аутосомные нарушения Синдром Дауна - трисомия 21 (Рис. 1) - наиболее изученная хромосомная болезнь. Частота синдрома Дауна среди новорождённых равна 1:700-1:800, не имеет какой-либо временной, этнической или географической разницы у родителей одинакового возраста. Частота рождения детей с синдромом Дауна зависит от возраста матери и в меньшей мере от возраста отца. Цитогенетические варианты синдрома Дауна разнообразны. Однако основную долю (94-95%) составляют случаи простой полной трисомии 21 как следствие нерасхождения хромосом в мейозе. При этом материнский вклад нерасхождения в эти гаметические формы болезни составляет 80%, а отцовский - только 20%. Причины такой разницы неясны. Небольшая (около 2%) доля детей с синдромом Дауна имеет мозаичные формы (47+21/46). Примерно 3-4% больных с синдромом Дауна имеют транслокационную форму трисомии по типу робертсоновских транслокаций между акроцентриками. Почти 50% транслокационных форм наследуется от родителей-носителей и 50% - транслокации, возникшие de novo. Соотношение мальчиков и девочек среди новорождённых с синдромом Дауна составляет 1:1. Большое значение для диагностики имеет динамика физического и умственного развития ребёнка. При синдроме Дауна и то и другое задерживается. Рост взрослых больных на 20 см ниже среднего. Задержка в умственном развитии достигает имбецильности, если не применяются специальные методы обучения. Дети с синдромом Дауна ласковые, внимательные, послушные, терпеливые при обучении. Коэффициент умственного развития (IQ) у разных детей широко варьирует (от 25 до 75). Реакция детей с синдромом Дауна на факторы окружающей среды часто патологическая в связи со слабым клеточным и гуморальным иммунитетом, снижением репарации ДНК, недостаточной выработкой пищеварительных ферментов, ограниченными компенсаторными возможностями всех систем. По этой причине дети с синдромом Дауна часто болеют пневмониями, тяжело переносят детские инфекции. У них отмечается недостаток массы тела, выражен авитаминоз. Врождённые пороки внутренних органов, сниженная приспособленность детей с синдромом Дауна часто приводят к летальному исходу в первые 5 лет. Следствием изменённого иммунитета и недостаточности репарационных систем (для повреждённой ДНК) являются лейкозы, часто встречающиеся у больных с синдромом Дауна. Синдром Эдварда (Рис. 2,3) - встречается редко. Синдром Эдвардса - хромосомное заболевание, обусловленное трисомией по 18-ой хромосоме и сопровождающееся множественными пороками развития. Для синдрома Эдвардса характерны своеобразные фенотипические признаки (долихоцефалическая форма черепа, микрофтальмия, недоразвитие ушных раковин, микроретрогнатия и др.), аномалии опорно-двигательной, сердечно-сосудистой, пищеварительной, мочеполовой системы, ЦНС. Синдром Эдвардса может быть диагностирован на этапе беременности (УЗИ-скрининг, инвазивная пренатальная диагностика) либо уже после рождения ребенка на основании внешних признаков и цитогенетического исследования. Дети с синдромом Эдвардса нуждаются в симптоматическом лечении и хорошем уходе. Большинство детей с Синдром Эдвардса умирает до достижения ими 1-летнего из возраста. Проблема гена случается наиболее часто случайно, хотя более вероятна у старших женщин, и семейное наследование очень редко. Смешанный вариант, известный как мозаицизм, который появляется, когда половина генов органа нормальна, а половина несет Т-18, ведя к менее серьезному изменению. Среди наиболее частых врождённых аномалий, ассоциированных с T-18: · гидроцефалия · дефект нервной трубки · кистозная гигрома · «скрюченные» пальцы рук · ступни рокера · дефекты межжелудочковой перегородки · микрогнатия · ступни-молоты · сжатые в кулаки руки · диафрагмальная грыжа · омфалоцеле · серьезное ограничение роста Синдром Патау (Рис. 4) - хромосомное заболевание, которое характеризуется наличием в клетках дополнительной хромосомы 13. Частота синдрома среди новорожденных составляет 1:7000. Оба пола поражаются с равной частотой. В 50% случаев беременность осложняется многоводьем. Масса при рождении значительно ниже нормы: часто в родах отмечается асфиксия (кислородное голодание). Клинические проявления: микроцефальнийчереп с низким скошенным лбом, вдавленными височными участками и дефектами кожи. Глазные щели узкие, расположены горизонтально, недоразвитые глазные яблоки, наблюдается помутнение роговицы. Нос сплюснутый, широкий, с впалой переносицей и тупым втянутым кончиком. Ушные раковины расположены очень низко, маленькие мочки прижаты к голове, завитки неправильной формы. Неправильное развитие кортиевого органа часто приводит к глухоте. Одним из основных признаков синдрома является «заячья губа» и «волчья пасть». Почти всегда присутствуют аномалии сердца и сосудов: дефект межжелудочковой и межпредсердной перегородок, незаращение артериального протока, патология клапанного аппарата, стеноз легочной артерии. Половина больных синдромом Патау имеют пороки развития почек и мочевыводних путей: кистозная почка, гидронефроз, плазия почек, удвоение почек и др. Довольно часто встречаются различные дефекты и аномалии положения органов пищеварения: аномалии кишечника, патологии брыжейки (дупликатура брюшины, посредством которой полые органы брюшной полости прикреплены к задней стенке живота), дивертикул Меккеля, кистозно-фиброзные изменения поджелудочной железы. У 50% девочек наблюдается удвоение влагалища и двурогая матка. Синдром кошачьего крика (Рис. 5) - это нарушение вызвано делецией короткого плеча 5 хромосомы. Мяуканье и звуки, подобные кошачьему крику типичны для этой патологии. Часто наблюдается отставание в умственном развитии и пороки сердца. Срок жизни таких больных незначительно выше, чем у пациентов с трисомией 18 и 13 хромосом. Заключение. Приложение Рисунок 1 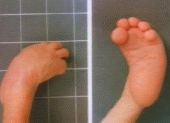 Рисунок 2  Рисунок 3  Рисунок 4  Рисунок 5 |
