|
|
статья. иммунка. Многие распространенные и редкие варианты, связанные с гематологическими признаками, были обнаружены путем вменения на крупномасштабных контрольных панелях
Резюме
Многие распространенные и редкие варианты, связанные с гематологическими признаками, были обнаружены путем вменения на крупномасштабных контрольных панелях. Однако большинство общегеномных ассоциативных исследований (GWASs) были проведены у европейцев, и определение причинно-следственных вариантов оказалось сложной задачей. Мы провели GWAS общего количества лейкоцитов, нейтрофилов, лимфоцитов, моноцитов, эозинофилов и базофилов, полученных из 109 563 748 вариантов в аутосомах и Х-хромосоме в Trans-Omics для точной медицины (TOPMed) программа, которая включала данные от 61 802 человек различного происхождения. Мы обнаружили и воспроизвели 7 ассоциаций лейкоцитарных признаков, включая (1) ассоциацию между хромосомой X, псевдоаутосомной областью (PAR), некодирующим вариантом, расположенным между генами рецепторов цитокинов (CSF2RA и CLRF2) и меньшим количеством эозинофилов; и (2) ассоциации между одиночными вариантами
, обнаруженными преимущественно среди Афроамериканцы по локусам S1PR3 (9q22.1) и HBB (11p15.4) и количеству моноцитов и лимфоцитов соответственно. Далее приводятся доказательства, указывающие на то, что недавно обнаруженный вариант хромосомы X PAR, снижающий уровень эозинофилов, может быть связан со сниженной восприимчивостью к распространенным аллергическим заболеваниям, таким как атопический дерматит и астма. Кроме того, мы обнаружили бремя очень редкие Варианты FLT3 (13q12.2), связанные с количеством моноцитов. В совокупности эти результаты подчеркивают полезность секвенирования всего генома в различных образцах для выявления ассоциаций, пропущенных GWASs, основанным на европейском происхождении.
Вступление
Количество циркулирующих белых кровяных телец (лейкоцитов) является важными клиническими параметрами, которые используются для мониторинга общей активности заболевания и толерантности к терапии онкологических и ревматологических заболеваний. Лейкоциты происходят из гемопоэтических стволовых клеток и в процессе дифференцировки разделяются на две различные линии: миелоидную (нейтрофилы, базофилы, эозинофилы и моноциты) и лимфоидную (лимфоциты). Изучая генетические детерминанты количества лейкоцитов, мы смогли получить более полное представление о кроветворении и сложной роли лейкоцитов как при остром, так и при хроническом воспалении.
Общее и дифференциальное количество лейкоцитов являются сложными, полигенными, количественными признаками, и генетический вклад в различия в количестве лейкоцитов (наследуемость) оценивается как
50%-60%. Многочисленные недавние исследования характеризовали как распространенные (частота минорных аллелей [MAF] более 5%), так и нечастые (MAF от 0,5% до 5%) вариации, способствующие подсчету WBC в европейских, африканских, восточноазиатских и испаноязычных популяциях. На сегодняшний день большинство исследований генетики подсчета WBC использовали комбинацию методов исследования, включая стандартные наборы генотипирования по всему геному, секвенирование экзома, генотипирование экзомных чипов, и применение общегеномного вменения с использованием эталонных панелей. Очевидным пробелом в этих методах исследования является всесторонний, общегеномный опрос распространенных и редких вариация, которую можно было бы упустить при использовании подходов, основанных на вменении.
Анализ, основанный на секвенировании всего генома (WGS), в значительной степени устраняет эти пробелы, особенно у лиц неевропейского происхождения. Важно отметить, что WGS может оценивать варианты, специфичные для популяции, включая варианты, которые часто плохо вменяются стандартными контрольными панелями и массивами генотипирования. Здесь мы использовали глубокие (303) данные WGS от 61 802 человек, включая афроамериканцев (AA), восточноазиатцев ((EAST), европейско-американские (EA) и испаноязычные/ латиноамериканские (HA) субъекты. Данные были получены в рамках Национального Институт сердца, легких и крови (NHLBI) - программа Trans-Omics for Precision Medicine (TOPMed), исследующая генетику количества лейкоцитов.
Материал и методы
Образцы TOPMed
Программа TOPMed NHLBI включает в себя несколько исследований для родителей. Исходные исследования, которые внесли свой вклад в наш анализ, включали риск атеросклероза в сообществах (ARIC), сложную болезнь амишей Исследовательская программа (Amish), Биобанк Биома (BioMe), Риск развития сердечно-сосудистых артерий у молодых людей (CARDIA), Исследование сердечно-сосудистого здоровья (CHS), Генетическая эпидемиология
COPD (COPDGene), Исследование сердца Фреймингема (FHS), Генетическое исследование риска атеросклероза (GeneSTAR), Исследование здоровья испаноязычного сообщества/Исследование латиноамериканцев (HCHS/SOL), Исследование сердца Джексона (JHS), Многоэтническое исследование атеросклероза (MESA), Семейное исследование сердца в Сан-Антонио (SAFS), и Инициатива по охране здоровья женщин (WHI). Дополнительная информация о структуре каждого исследования и выборке лиц в каждой когорте для WGS доступна в дополнительной информации. Участники, включенные в эти анализы (уникальный n = 61 865), показаны в таблице S1, стратифицированные по исследованию, группе происхождения (см. Дополнительные методы) и признаку WBC. Для этих анализов 1% участников - азиаты, 23% - чернокожие, 22% - испаноязычные и 54% белые. Все исследования были одобрены соответствующими институциональными наблюдательными советами (IRBS), и от всех участников было получено информированное согласие.
TOPMed WGS и контроль качества
WGS была выполнена на средней глубине 383 в шести центрах секвенирования (Broad Genomics, Northwest Genome Center, Illumina, Нью-Йоркский центр генома, Бэйлор и Институт генома Макдоннелла) с технологией Illumina X10 и ДНК из крови. Здесь мы сообщаем об анализе из набора данных «Freeze 8», где считывания были согласованы с построением генома человека GRCh38 с помощью общего конвейера во всех центрах секвенирования. Чтобы выполнить контроль качества вариантов (QC) в наборе данных Freeze 8, мы обучили классификатор машины опорных векторов (SVM) на известные сайты вариантов (положительные метки) и менделевские несогласованные варианты
(отрицательные метки). Дальнейшая фильтрация вариантов была проведена для вариантов с избыточной гетерозиготностью и менделевским диссонансом. Образец Меры контроля качества включали соответствие между аннотированным и предполагаемым генетическим полом; соответствие между данными о генотипе предыдущего массива и данными TOPMed WGS; и проверку родословной. Подробности, касающиеся «замораживания» генотипа, лабораторных методов, обработки данных и контроля качества, описаны на веб-сайте TOPMed и в общем документе, сопровождающем регистрационный номер dbGaP для каждого исследования.
Измерения фенотипа WBC и критерии исключения
Лейкоциты, базофилы, эозинофилы, нейтрофилы, лимфоциты и моноциты подсчитывали в подгруппе образцов TOPMed freeze 8 (таблица S1) с помощью автоматизированных клинических гематологических анализаторов. Каждый из фенотипов определяется как концентрация типа клеток в крови и измеряется в миллиардах/литр. QC, зависящий от конкретного признака, исключил участников со значениями количества лейкоцитов > 100*109 клеток/л (n = 5), значения нейтрофилов > 75 * 109 клеток/л (n = 1), значения моноцитов > 15 * 109 клеток/л (n = 1), значения лимфоцитов > 150* 109 клеток/л (n = 1), показатели эозинофилов > 20 * 109 клеток/л (n = 1), а значения базофилов равны 0,9 * 109 клеток/л (n = 1). Кроме того, в тех случаях, когда было доступно несколько измерений, мы сохраняли только одно измерение для каждого индивидуума и каждой черты.
Одновариантные ассоциативные тесты для количественных признаков
Мы провели общегеномные одновариантные ассоциативные тесты с использованием двухэтапной линейной смешанной модели (LMM). На первом этапе мы подогнали «нулевую модель» под нулевую гипотезу об отсутствии генетической ассоциации и не включили генетические варианты в модель. Мы включили пол, возраст, комбинированное исследование по фазовым переменным (например, WHI_2 относится ко 2-й фазе исследования WHI), а первые 11 PC-Air основные компоненты (PCs) генетического происхождения как фиксированные эффекты.
Чтобы учесть генетическое родство, мы включили разреженную эмпирическую матрицу родства 4-й степени (KM), вычисленная с помощью PC-Relate. Чтобы лучше контролировать геномную инфляцию, мы допустили гетероскедастичность в отклонениях ошибок путем моделирования отдельных компонентов остаточной дисперсии, по одному для каждого исследования, по группам происхождения (например, WHI_White). Подробная информация об оценке группы предков приведена в дополнительных методах. Чтобы повысить мощность и соответствующим образом контролировать ошибку типа I в настройках с ненормальным распределением фенотипов, мы использовали
полностью скорректированный двухэтапный подход для подгонки нулевой модели. В этап 1, мы сопоставляем LMM с наблюдаемыми значениями фенотипа в качестве результата, фиксированными эффектами в качестве ковариат, разреженным KM и гетерогенными остаточными отклонениями. Мы применили основанное на ранге обратнонормальное преобразование к остаткам от результатов этапа 1, а затем изменил их масштаб на исходную дисперсию. На этапе 2 мы подгоняем другой LMM, используя масштабированные остатки, полученные на этапе
1 в качестве результата и с использованием тех же ковариат, того же КМ и той же гетерогенной модели остаточной дисперсии, что и на этапе 1.
Наконец, мы использовали результаты этапа 2 в качестве характеристики, представляющей интерес для выполнения балльного теста генетической ассоциации. В ассоциативный анализ мы включили варианты, которые имели количество минорных аллелей (MAC) не менее 5, прошли фильтры качества TOP Med Informatics Research Center (IRC) и имели менее 10% образцов с глубиной считывания последовательности менее 10. Для определения статистической значимости использовался пороговый уровень 5 * 10-8.
Одновариантные ассоциативные тесты для определения количества базофилов как бинарного признака
Вместо тестирования базофилов как непрерывного признака мы провели общегеномные тесты на одно-вариантную ассоциацию базофилов как бинарного признака, дихотомизированного при 0,05 * 109 клеток/л (basophil3 R 0,05 против basophil < 0,05). Мы применили обобщенную линейную смешанную модель (GLMM) с биномиальным семейством и логит-линком с помощью штрафного квази-правдоподобия подхода GMMAT, поскольку наш результат больше не был количественным. Были включены те же ковариаты с фиксированным эффектом и разреженные КМ, что и для анализа количественных признаков. Поскольку дисперсионная модель для GLMM определяется биномиальным семейством и функцией связи, мы не использовали гетерогенные группы остаточных дисперсий или двухэтапную процедуру нормализации ранга. Мы провели общегеномные ассоциативные тесты, основанные на статистике баллов и аппроксимации значений p в точке седла (SPA). Было показано, что метод SPA лучше контролирует ошибку типа I, даже когда соотношение пораженных и контрольных особей несбалансировано, например, при тестировании низкочастотных и редких вариантов, когда количество носителей намного меньше размера выборки.
Условный анализ
Мы выполнили условные одновариантные ассоциативные тесты, в которых, в дополнение к поправке на ковариаты с фиксированным эффектом и разреженные КМ, которые использовались в одновариантных анализах, мы сделали поправку на варианты, ранее известные как связанные с результатами (таблица S3). Сначала мы сопоставили известные варианты с вариантами TOPMed на основе положения и аллелей и выбрали варианты, которые прошли фильтры качества TOPMed IRC. Затем мы использовали неравновесное сцепление генов (LD) (с порогом R2 > 0,8), чтобы сократить набор совпадающих вариантов для каждого признака отдельно и проверили коллинеарность сокращенных вариантов с ковариатами. Окончательный набор вариантов был включен в первый раунд условного анализа. После первого раунда мы проверили, были ли оставшиеся значимые варианты близки (в пределах окна размером 1 Мб) к известным вариантам, которые не прошли IRC-фильтры TOPMed. Эти варианты, в дополнение к набору вариантов из первого раунда, были включены во второй раунд условного анализа.
Основанные на генах совокупные тесты на редкие варианты
Чтобы повысить способность обнаруживать ассоциации с редкими вариантами, мы внедрили несколько стратегий агрегирования вариантов и тестирования на кумулятивные ассоциации группировок на основе генов с признаками. Мы реализовали в общей сложности пять стратегий группирования вариантов: три стратегии включали только кодирующие варианты, а две стратегии включали кодирующие и некодирующие варианты из областей энхансера (Энхансеры - это области генома, которые являются основными регуляторными элементами генов) и промотора, но только те, которые имеют «вредные» последствия для соответствующего гена, «вредные» определяются различными фильтрами на основе аннотаций; подробная информация приведена в дополнительных методах. Мы выполнили агрегированные тесты, используя эффективный тест ассоциации смешанных моделей с набором вариантов (SMMAT), который более эффективен в вычислительном отношении, чем SKAT-O (оптимизированный SKAT [тест ассоциации ядра последовательности]) и более мощный, чем тесты нагрузки или только SKAT. В тесте SMMAT использовалась та же нулевая модель, которая подходила для одновариантных анализов, и значение p было построено из комбинации значения p нагрузки смешанной модели с асимптотически независимым скорректированным SKAT-подобным
значением p по методу Фишера. В наш анализ мы включили немономорфные варианты, которые имели MAF менее 1% и которые прошли те же качественные фильтры, которые использовались для одновариантных анализов. Чтобы увеличить вес более редких вариантов, мы использовали веса, основанные на MAF и заданные бета-распределением с параметрами 1 и 25. Мы определили статистическую значимость, используя поправку Бонферрони для количества агрегированных групп, протестированных в каждой стратегии агрегирования.
Анализ пропорций подтипов WBC
В дополнение к анализу количества подтипов WBC, мы также проанализировали пропорции подтипов WBC для всех реплицированных, статистически значимых сигналов подсчета подтипов WBC. Для этого мы идентифицировали образцы, у которых количество лейкоцитов и соответствующее количество подтипов лейкоцитов были собраны при одном посещении, и разделили количество подтипов лейкоцитов на общее измеренное количество лейкоцитов. Мы исключили образцы, в которых соотношение количества WBC-подтипов к количеству WBC было больше 1. Эта пропорция рассматривалась как фенотип и моделировалась аналогично другим фенотипам, т.е. с помощью двухэтапных пленок, описанных выше.
Анализ точного отображения
После условного анализа мы провели статистическое точное картирование, используя следующий подход: поскольку наши условные анализы включали один независимый вариант в каждом локусе, мы предположили один причинно-следственный вариант в каждом локусе. Затем мы адаптировали метод, предложенный Мюллером и др., чтобы присвоить каждому варианту вероятности последующего включения (PIPs) и построить 95% достоверные наборы. Вкратце, мы рассмотрели все варианты в пределах выше и ниже 250 кб по потоку от sentinel SNP и преобразовали сводную статистику в приблизительные коэффициенты Байеса (aBFs) следующим образом:
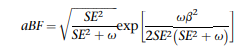
где β и SE - размер эффекта варианта и стандартная ошибка, соответственно, и ω представляет собой предшествующую дисперсию аллельных эффектов. Как и в Maller и др., мы устанавливаем ω = 0,04. Затем мы рассчитали PIP каждого варианта путем деления aBF варианта на сумму aBFs для всех вариантов в локусе. Мы сгенерировали 95% достоверных наборов, упорядочив все варианты (в определенном местоположении) от наибольшего до наименьшего PIP и включая варианты до тех пор, пока совокупные пункты PIPs ≥ 0,95.
Анализ гаплотипов
На основе результатов анализа условных единичных вариантов мы провели анализ гаплотипов для бета-гемоглобина (HBB) области (rs334 и rs33930165) в общем количестве лейкоцитов и лимфоцитов NRIP1 области (rs28574812 и rs2823002) в моноцитах и общем количестве лейкоцитов. Мы сконструировали гаплотипы 2-SNP на основе данных поэтапного генотипа и идентифицировали гаплотипы с ненулевыми частотами. Мы подсчитали количество копий каждого гаплотипа у каждого субъекта и включили количество копий каждого нереферентного гаплотипа в качестве ковариат в модель. Эталонным гаплотипом считался гаплотип с наибольшей частотой. Используя нулевую модель из анализа одного варианта, мы провели ассоциативные тесты и сообщили о результатах, специфичных для гаплотипа.
Анализ PheWAS
Мы извлекли результаты общефеномного ассоциативного сканирования (PheWAS) для семи новых реплицированных сигналов из UKBiobank (UKBBB) и BioVU biobank. Результаты UKBB были получены из UKBB ICD PheWeb, размещенного в Мичиганском университете, на основе 408 961 образца от белых британских участников. Мы рассмотрели 1261 phecode с по меньшей мере 100 затронутыми людьми и соответствующим порогом с поправкой Бонферрони для значимости 0,05/1261 = 3,96* 10-5. BioVU - это биобанк Медицинского центра Университета Вандербильта (VUMC), в котором хранятся деидентифицированные образцы ДНК, связанные с фенотипическими данными, полученными из системы электронной медицинской карты (EHR) VUMC. Запросы PheWAS
в BioVU были ограничены афроамериканцами (n » 5000). Для поиска rs334 у нас был доступ к образцам из 14 000 афроамериканцев, которые были либо гетерозиготными в rs334 или имели две копии эталонного аллеля. Фенотипы были получены из биллинг кодов (код (медицинской) услуги для оплаты) EHRS. Связь между каждым бинарным phecode и SNP оценивалась с использованием логистической регрессии с поправкой на ковариаты возраста, пола, партии генотипирующего массива и 10 основных компонентов родословной. Мы рассмотрели 726 phecodes по меньшей мере в 100 случаях с соответствующим порогом значимости с поправкой на Бонферрони, равным 0,05/726 = 6,89*10-5.
Анализ UKBiobank астмы, COPD и атопического дерматита с помощью rs28532112
Мы сконструировали фенотипы хронической обструктивной болезни легких (ХОБЛ= COPD), астмы и атопического дерматита (AD), как определено в Wu и др., используя коды ICD10 для выбора группы пациентов и контрольной группы. Первоначально отобранный набор затронутых лиц и контрольная группа была очищена от родства путем удаления одного члена каждой связанной пары итеративным способом до тех пор, пока не осталось ни одного родственного субъекта. Используя оставшуюся группу пострадавших лиц и оставшийся пул контрольных лиц, мы отобрали фиксированное количество контрольных лиц для каждого пострадавшего лица, соответствующих полу, возрасту и происхождению. Фиксированное число, используемое для соотношения контрольных особей к пораженным особям, было скорректировано, чтобы получить общее число n в диапазоне от 40 000 до 80 000 субъектов. Анализ ассоциаций был проведен с помощью OASIS pipeline.
TOPMed анализ rs28532112 с астмой и тяжестью астмы
TOPMed сгенерировал WGS для n = 869 пациентов со статусом астмы (410 астматиков и 459 неастматических лиц) для Барбадосского исследования генетики астмы (BAGS) и n = 611 пациентов с астмой из исследования Программы исследований тяжелой астмы (SARP). Мы использовали GENESIS для проведения ассоциативных тестов для rs28532112 с астмой (в пакетах) и включили возраст, партию и пол в качестве ковариат. Мы также провели тесты на связь с тяжестью астмы (в остром состоянии), измеренной по объему предварительного форсированного выдоха 1 (преОФВ1), для rs28532112; мы контролировали возраст, пол и индекс массы тела и стратифицировали по происхождению (EA [n = 218] и AA [n = 393]).
Анализ ADRN rs28532112
Чтобы определить генетические факторы риска БА (бронхиальная астма), мы провели WGS у 777 пациентов из Исследовательской сети по атопическому дерматиту (ADRN) Национального института аллергии и инфекционных заболеваний, как описано ранее. Это включает 237 незатронутых индивидуумов, 491 индивидуума, страдающего БА без герпетической экземы, и 49 индивидуумов, страдающих БА с герпетической экземой. Чтобы выполнить ассоциативные тесты между rs28532112 и AD, мы сравнили 491 человека, пораженного AD, с 237 людьми, не затронутыми AD, используя обобщенную логистическую регрессию (GLM) и PLINK /Seq и корректировку для первых пяти PCs в качестве ковариат.
TOPMed анализ rs28532112 с ХОБЛ и функцией легких
Подробную информацию об анализе TOPMed ХОБЛ и функции легких можно найти в Zhao и соавт. Вкратце, в анализе приняли участие 19 996 человек из разных этнических групп, в том числе 12 314 восточных, 6450 афроамериканцев и 1232 образца, классифицированных как «другие» от TOPMed. Гармонизация фенотипа показателей теста функции легких, включая ОФВ1 до введения бронходилататора, форсированную жизненную емкость (ФЖЕЛ) и соотношение ОФВ1:ФЖЕЛ, проводилась в соответствии со стандартными протоколами. Мы включили ковариационную поправку на возраст, пол, рост, вес (только для ФЖЕЛ), исследование, курящие, курившие, количество лет курения, первые 10 PCs родословной и центр секвенирования в рамках LMM, чтобы учесть гетерогенную дисперсию между исследованиями с использованием GENESIS. Анализ "случай-контроль" включал ковариационную поправку на возраст, пол, учебу, количество лет, независимо от того, курил ли человек когда-либо или никогда не курил, первые 10 PCs родословной и центр секвенирования.
Протеомное профилирование SOMAScan и анализ pQTL rs334
У участников JHS и MESA TOPMed образцы плазмы EDTA, собранные при соответствующих базовых обследованиях и хранящиеся в морозильных камерах при температуре -70 °C, были подвергнуты протеомным измерениям с помощью SOMAscan, платформы протеомики на основе одноцепочечных ДНК-аптамеров, содержащей 1305 аптамеров. В JHS образцы (n = 2054 AA) были обработаны тремя отдельными партиями. Белки определяли количественно в относительных флуоресцентных единицах, концентрация которых пропорциональна концентрации белка в образце плазмы. Протеомные измерения были стандартизированы для набора контрольных образцов (объединенной плазмы), содержащихся в каждом 96-луночном планшете, и полученные значения были логарифмически преобразованы и масштабированы до среднего значения 0 и стандартного отклонения 1. Связь между генотипом rs334 и значениями белка оценивалась с помощью линейных моделей смешанных эффектов. В JHS белки были стандартизированы внутри каждой партии, а затем обратно нормализованы по партиям и скорректированы с учетом возраста, пола и партии. Образцы MESA (n = 189 АА и 301 НА) были скорректированы с учетом возраста, пол, этнической принадлежности (латиноамериканец да или нет), номер телефона и сайт. Результаты, относящиеся к конкретной группе людей, были подвергнуты мета-анализу с помощью взвешивания с обратной дисперсией. Использовался порог значимости, скорректированный по Бонферрони, равный 3,8* 10-5 (0,05/1,301).
rs28532112 анализ ассоциации плазмы с белками
Далее мы провели количественный анализ белков плазмы целевого генотипа, чтобы определить ассоциацию rs28532112 с концентрациями интерлейкина-3 (IL-3), растворимого IL 3R-альфа, гранулоцитарно-макрофагального колониестимулирующего фактора (GM-CSF) и рецептора и лиганда тимусного стромального лимфопоэтина (TSLP) в плазме крови, используя доступные данные SOMAscan из 2544 многоэтнических образцов JHS и MESA TOPMed. Для определения растворимого альфа-рецептора GM-CSF мы использовали отдельную панель анализа близости неврологической связи (PEA), измеренную в 1328 многоэтнических выборках TOPMed WHI, скорректированных по возрасту и происхождению.
rs334 и расчетный анализ подмножества лимфоцитов в JHS
Данные массива Illumina MethylationEPIC (содержащий более 850 000 сайтов метилирования CpG) из n = 1756 участников JHS были получены из образцов крови, собранных во время базового обследования JHS. Уровни метилирования определяли количественно в терминах значения b, для которого в качестве отношения флуоресцентных сигналов использовали отношение интенсивностей между метилированным и неметилированным аллелем. Значения метилирования были нормализованы по отношению к интенсивности фонового цвета с помощью метода нормально-экспоненциального внеполосного анализа (NOOB). Количество клеток (гранулоциты, моноциты, естественные киллеры [NK], CD4+ Т-лимфоциты, наивные CD8+ Т-лимфоциты, истощенные цитотоксические CD8+ Т-клетки [определены как CD8-положительные, CD28 отрицательный, и CD45R отрицательный], и плазмобласты) оценивали в соответствии с методом Хаусмана и Хорвата и др. Связь между оцененным количеством клеток и носителями rs334 (исключая гомозиготы rs334), скорректированную на возраст, пол и 10 PCs генетического происхождения, оценивали с помощью обобщенного оценивающие уравнения в SAS 9.3, чтобы учитывалась семейная корреляция. |
|
|
 Скачать 55.9 Kb.
Скачать 55.9 Kb.