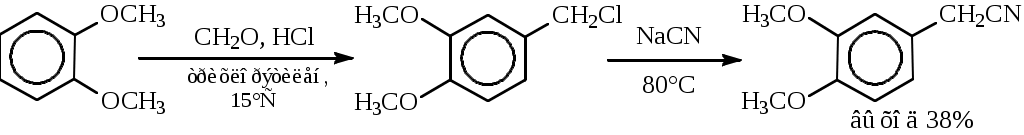
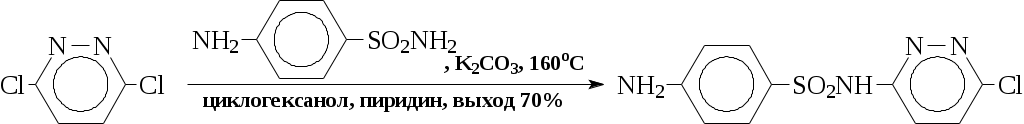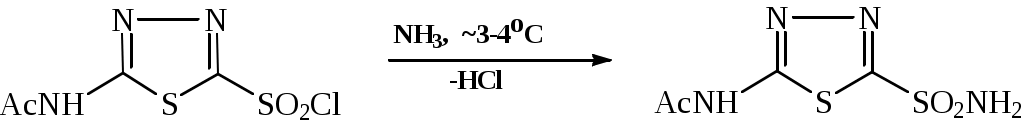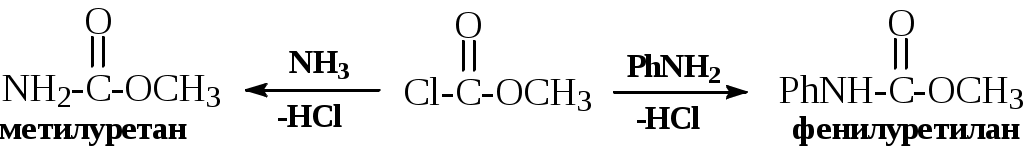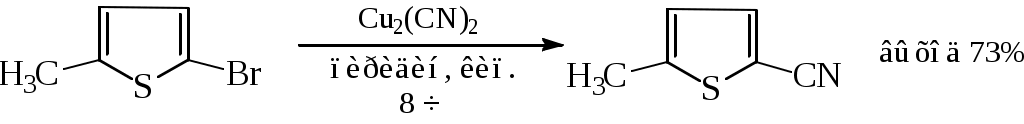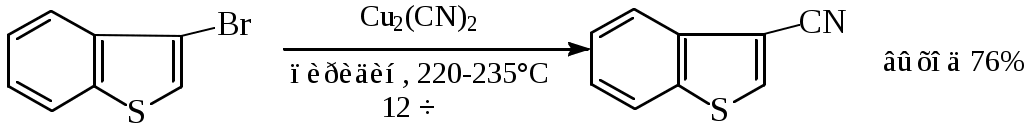ОПХСЛк9-11Замещение. Процессы замещения функциональных групп в молекуле органического соединения Нуклеофильное замещение галогена
 Скачать 0.52 Mb. Скачать 0.52 Mb.
|

1 2
5% молн.) иодида натрия. 2. В случае инертных галогенидов - 70-90%-й спирт или триэтиленгликоль. 3. Наилучшие результаты дают биполярные апротонные растворители (например, диметилсульфоксид, диметилформамид).
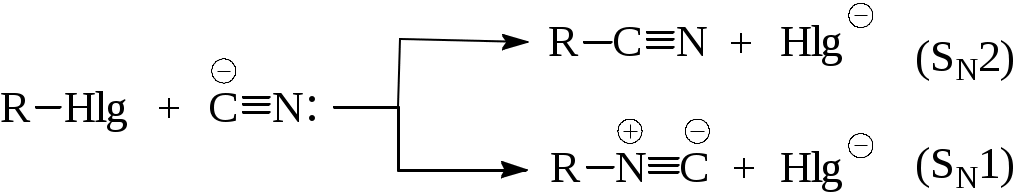
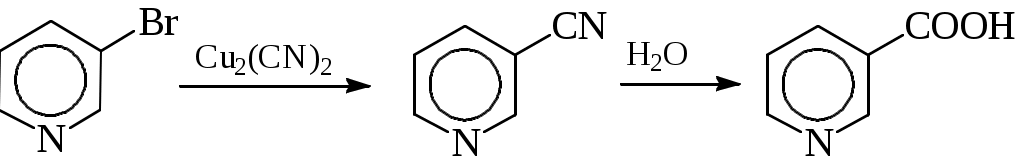
на 100оС), увеличить селективность процесса и выход продукта. Предполагают, что реакция идет через стадию образования медь-органических комплексов: Процессы замещения функциональных групп в молекуле органического соединенияНуклеофильное замещение галогена- позволяет получать представители практически всех классов органических соединений (спирты, эфиры, амины, нитрилы и др.), поэтому эти реакции широко применяются в синтезе лекарственных веществ. 1.1. Основные механизмы реакцийЗамещение галогена у sp3-гибридного атома углерода может осуществляться как по SN1, так и по SN2 механизмам. Замещение галогена у sp2-гибридного атома углерода (в арил- и винилгалогенидах) идет либо по типу присоединения-отщепления, либо по типу отщепления-присоединения и значительно труднее, чем у sp3-гибридного. - SN1 механизм включает две стадии: а) диссоциация алкилгалогенида на ионы; б) взаимодействие катиона с нуклеофилом: 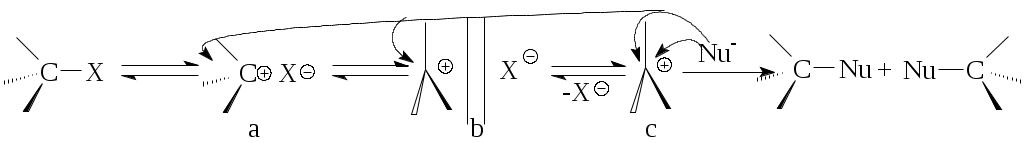 - Молекула алкилгалогенида может диссоциировать с последовательным образованием контактной ионной пары (а), сольватно-разделенной ионной пары (b) и сольватированных ионов (с). Нуклеофильная атака контактной ионной пары, в которой асимметрия в значительной мере сохраняется, приводит к обращению конфигурации. В сольватно-разделенной ионной паре одна сторона катиона экранируется сольватированным галогенид-ионом и атака нуклеофила более вероятна с другой стороны, что приводит к преимущественному обращению конфигурации, но селективность снижается, и рацемизация увеличивается. Полная рацемизация возможна лишь при образовании свободного катиона (с). Однако, полная рацемизация для оптически активных галогенидов при механизме SN1 обычно не наблюдается. Рацемизация составляет от 5 до 20%, следовательно, сольватированный катион практически не образуется. - Стадия образования карбокатиона является лимитирующей, а, следовательно, стабильность катиона определяет быстроту прохождения процесса. Скоростьпроцессазависит также от концентрации алкилгалогенида и не зависит от концентрации нуклеофила. - Образование карбокатиона может являться причиной ряда побочных процессов: изомеризация углеродной цепи, элиминирование (EI) и др.: 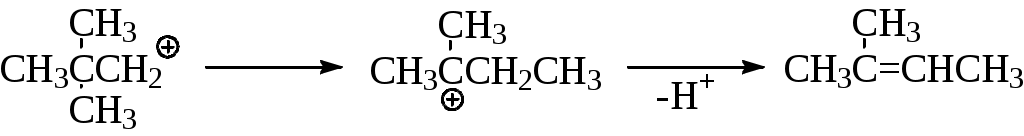 - Бимолекулярное замещение SN2 - синхронный процесс: - Нуклеофил Nu- атакует субстрат со стороны, противоположной уходящей группе. При этом реакция идет в одну стадию с образованием переходного состояния, в котором sp3-гибридизация центрального атома углерода изменяется на sp2- с р-орбиталью, перпендикулярной плоскости расположения гибридных орбиталей. Одна доля этой р-орбитали перекрывается с нуклеофилом, а вторая – с уходящей группой. Связь С-Nu образуется одновременно с разрывом связи С-Y. - Скорость превращения исходных веществв продукты реакции зависит: 1)от величины положительного заряда на атоме углерода субстрата, 2)пространственных факторов, 3)силы нуклеофила и 4)в кинетической областиот концентрации как нуклеофила, так и алкилгалогенида. При большом избытке нуклеофила реакция может протекать по первому или дробному порядку. (Термины SN1 и SN2 указывают лишь на молекулярность, но не на порядок реакции.) - Реакция всегда сопровождается обращением конфигурации. Побочной может быть реакция элиминирования Е2. - Механизм SNAr (присоединение - отщепление) - обычно реализуется при наличие электроноакцепторных заместителей, которые создают + (направляют нуклеофил) и стабилизируют -комплекс. В гетероциклах их роль выполняет гетероатом. В отличие от механизма SN2 для алкилгалогенидов нуклеофил образует новую связь раньше, чем рвется старая. 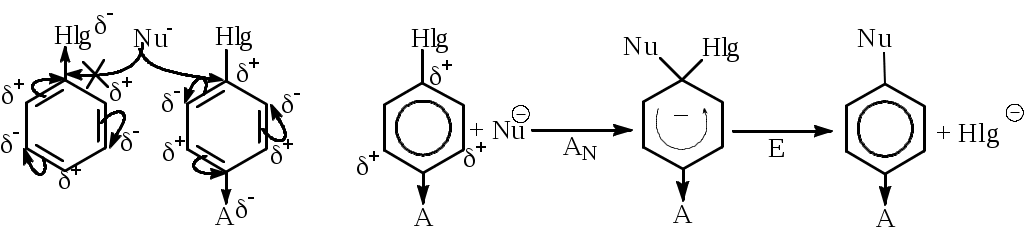  Структура (г) - очень устойчива, напоминает анион ациформы нитросоединения. Существование таких -комплексов (соли Мейзенгеймера 1902 г.) доказано экспериментально (ЯМР и рентгеноструктурный анализ). - Первая стадия а, следовательно, стабильность -комплекса, обычно, определяет скорость всей реакции. - Механизм отщепления – присоединения (SNEA) реализуется при замене галогена в галогенидах, не содержащих электроноакцепторных групп:   С помощью меченого атома углерода, а также реакциями с замещенными галогенбензолами, было показано, что в этом случае замещающая группа становится не только к тому атому углерода, где был галоген, но в равной степени и к соседнему атому, что объясняется образованием 1,2-дегидробензола. Образование дегидробензола было доказано как физико-химическими, так и чисто химическими методами. Так, при действии амальгамы лития на 1-фтор-2-бромбензол в присутствии диенофилов образующийся 1,2-дегидробензол вступает с ними в реакцию Дильса-Альдера:  1.2. Основные факторы, влияющие на ход процессаУсловия проведения и ход реакций замены галогена зависят от строения субстрата и реагента, полярности среды и природы уходящего галогена. 1.Строение субстратаА. Структура реагирующего соединения определяет механизм замещения галогена(напр., SN1 или SN2; SNAr(AE) или SNAr(EA)).  - Cувеличением разветвленности алифатического радикала в галогенидах создаются стерические препятствия для прямой атаки нуклеофила и увеличивается стабильность промежуточного карбкатиона, поэтому при переходе от первичного алкилгалогенида к третичному, в одних и тех же условиях механизм реакции может измениться от бимолекулярного до мономолекулярного. Этот процесс не является резким и зависит от ряда конкретных условий. Принципиально возможно протекание реакции по двум механизмам одновременно. - Аллил- и бензилгалогенидылегко реагируют как по SN1, так и по SN2 механизму. Однако, независимо от разветвленности радикала они образуют очень устойчивые карбокатионы, поэтому преимущественно реализуется SN1-механизм. При этом наблюдается аллильная перегруппировка, так как промежуточный аллилкатион может существовать в двух структурах:   В случае механизма SN2 перегруппировка наблюдается только в неполярном растворителе при взаимодействии сильного нуклеофила с аллилгалогенидом, у которого подход к атому углерода при галогене стерически затруднен:  - В ароматических галогенидах наличие электроноакцепторных заместителей в орто-, пара-положениях способствует SNArзамещению, а электронодонорных – механизму SNEA отщепления-присоединения, через дегидробензол. 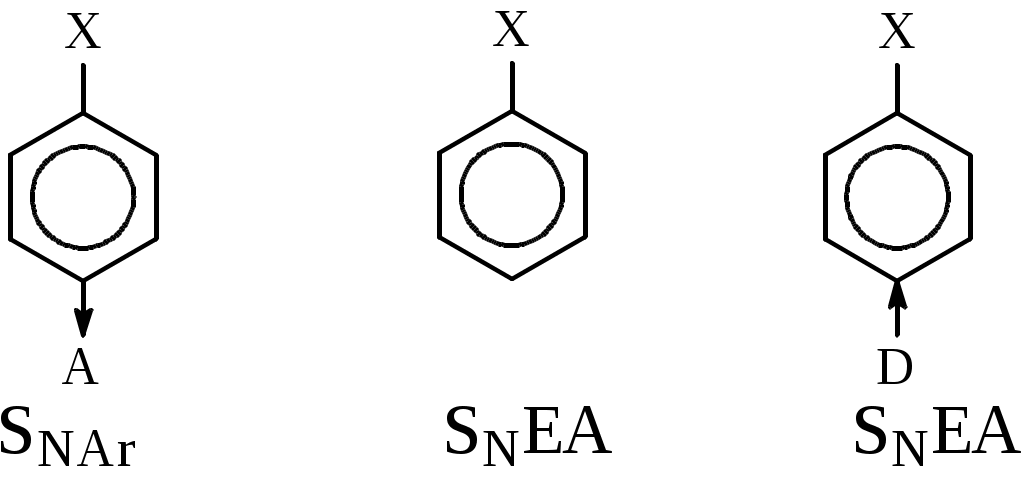 Б. Структура реагирующего соединения определяет и скорость процессов:  1. Скорости SN1 реакций алкилгалогенидов возрастают в ряду от метильного радикала к третичному, аллильному и бензильному. Находящиеся в -положении к реакционному центру предельные, фенильные и винильные радикалы, а также атомы, имеющие неподелённую пару электронов, за счет эффектов индукционного и сопряжения способствуют распределению электронного облака частицы, стабилизируют катион и ускоряют реакцию. При этом по силе активации один -фенильный радикал соответствует примерно двум алкильным заместителям. 2. Скорости SN2 реакций алкилгалогенидов возрастают в прямо противоположном направлении, наблюдаемому при SN1 замещении, если не учитывать повышенную активность первичных аллил- и бензилгалогенидов. Метильные и первичные галогениды реагируют очень гладко, вторичные – значительно хуже, а третичные часто не реагируют вообще, что объясняется, в основном, пространственными препятствиями для атаки нуклеофила, которые играют в SN2 замещении важную роль. 3. В ароматических галогенидах электроноакцепторныезаместители в орто-, пара-положениях существенно облегчают реакцию SNAr замещения, электронодонорные – затрудняют ее. Пространственные факторы при нуклеофильном замещении в ароматическом ряду не являются определяющими, так как атака направлена сбоку к плоскости ароматического ядра. 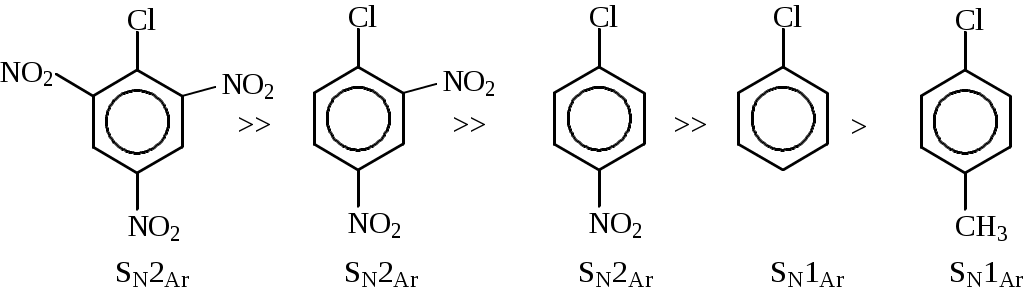 Так, 2,4,6-тринитрогалогенбензолы реагируют с водой, аммиаком и др. как хлорангидрид кислоты (очень легко!). Динитрогалогенбензолы реагируют с подобными реагентами медленнее. Замена галогена в о- и п-хлорнитробензоле проходит в щелочном растворе при 130-150°С, а хлорбензол гидролизуется до фенола лишь при температуре 350-400°С и давлении выше 30 МПа под действием 5% раствора щелочи и по другому механизму. Активирующее действие групп при одинаковом их размещении относительно галогена изменяется в следующем порядке: N2 > NO2 |
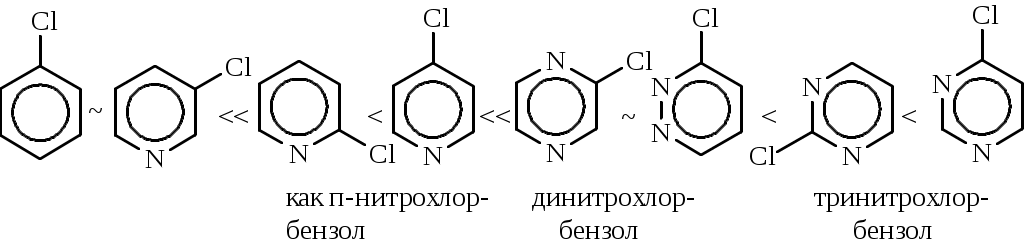
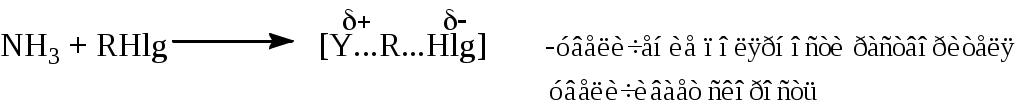
1.3. Замена атома галогена на –ОН- группу
1. Гидролиз моногалогенидов в синтетических целяхиспользуется редко, так как сами алкилгалогениды обычно получают из спиртов.
- Водойгидролизуют лишь галогениды с повышенной реакционной способностью. Аллил- и бензилхлориды превращаются в соответствующие спирты при кипячении в избытке воды. В остальных случаев даже при нагревании реакция идет очень медленно, т.к. вода является слабым нуклеофилом, а реакция алкилгалогенида с водой - обратимой:
- Водные растворы гидроксидов щелочных металлов или гидроксид серебра (суспензия оксида серебра в воде) являются более активными нуклеофильными реагентами и применяются в большинстве случаев:
- Однако в некоторых случаев для гидролиза используются даже водные растворы кислот. Например, в синтезе оригинального отечественного противоопухолевого препарата допан замену галогена в гетероароматическом ядре проводят при кипячении с соляной кислотой. Следует обратить внимание на то, что хлор в хлорэтильных группах при этом не гидролизуется. Выход очищенного продукта 85-86%.

- Для гомогенизации реакционной массы часто используют спирто-водную или водно-ацетоновую среды, так как алкилгалогениды нерастворимы в воде.
В зависимости от реагентов и условий проведения реакция может протекать как по SN1 так и по SN2-механизму, а также SNAr.
В зависимости от структуры субстрата и условий проведения реакции могут протекать вторичные процессы. Так, в синтезе мезатона и левомицетина, при щелочном гидролизе соответствующих галогенидов образуются окись м(п)-нитростирола или п-нитро--метоксистирол:


2. Гидролиз геминальных дигалогенпроизводных, у которых оба атома галогена связаны с одним атомом углерода, приводит к образованию альдегидов или кетонов. Так получают, например, бензофенон:
- Омыление проводят в слабощелочной среде в водных растворах карбонатов, ацетатов, формиатов или оксалатов натрия или калия, так как в сильнощелочных средах возможны побочные процессы: альдольная, кротоновая конденсация, окисление альдегидов и др.
- Бензилиденхлориды и бензилиденбромиды гладко гидролизуются до бензальдегидов под действием концентрированной серной кислоты. Электронодонорные группы в ядре облегчают гидролиз, электроноакцепторные – затрудняют. В последнем случае следует повысить температуру реакции до 120-130°С (но не выше, т.к. при более высокой температуре альдегиды будут интенсивно окисляться серной кислотой).
3. Гидролиз соединений, содержащих три атома галогена у одного атома углерода, приводит к образованию карбоновых кислот, однако тригалогениды трудно доступны и практическое применение метода ограничено:
При проведении гидролиза в присутствии спирта можно сразу получить эфир кислоты.
Хлороформ в присутствии основания гидролизуется быстрее, чем дихлорметан или тетрахлорметан и образует не только муравьиную кислоту, но и монооксид углерода. Считают, что реакция протекает через стадию образования дихлоркарбена:
Дихлоркарбен очень активная частица. Её образование подтверждается целым рядом реакций. Так, например, в присутствии пиррола дихлоркарбен присоединяется к гетероциклу с последующей перегруппировкой в 3-хлорпиридин:

4. При гидролизе CCl4образуется фосген:
1.4. Замена атома галогена на алкокси- или феноксигруппу (–OR,-OAr)
- образование простых эфиров - в синтезе лекарственных препаратов используется значительно чаще, чем гидролиз.
В качестве реагента используют либо алкоголят, либо спирт в щелочной среде. В случае фенольных нуклеофилов (ArOH) используют катализатор - соли меди, при этом считают, что активным реагентом является ArOCu. Выходы эфиров по этому методу обычно высокие.
Ароматические субстраты (арилгалогениды) должны быть активированными, иначе выход целевого продукта (эфира) может оказаться низким за счет побочных процессов.
1) При получении сульфапиридазина 3-хлор-6-сульфаниламидо-пиридазин нагревают 9 ч при 130°С и давлении 0,7-0,8 МПа с большим избытком едкого кали в среде метанола:

2) В синтезе сульфадиметоксина замена хлора в 4-амино-2,6-дихлорпиримидине происходит при 20-часовом кипячении в метанольном растворе NaOH:

3) В ряду пятичленных гетероароматических соединений такие реакции проводят лишь при наличие электроноакцепторных заместителей:

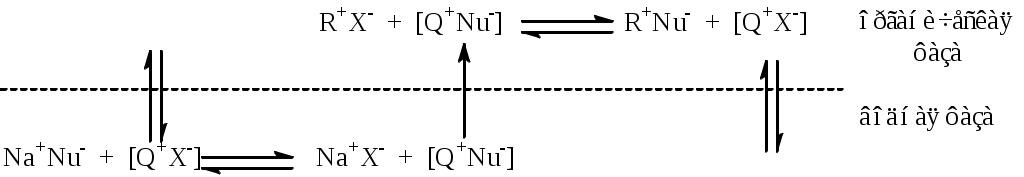
Применение метода межфазного катализа в синтезе простых эфиров позволяет повысить выход продукта, скорость реакции и технически упростить процесс.
При получении эфиров по Вильямсону в большинстве случаев используют обезвоженные реагенты и растворители, а также такие сильные основания как металлический натрий и амид натрия, необходимые для получения алкоголята. Это усложняет производство и повышает его опасность.
В двухфазном синтезе в качестве основания используется концентрированный водный раствор щелочи (обычно 50%-ный) и не надо обезвоживать растворители. Щелочь депротонирует спирт в водной растворе или на границе раздела фаз. Алкоголят - ион образует ионную пару с липофильным катионом межфазного катализатора (чаще всего четвертичной соли аммония) и переходит в органическую фазу. Гидроксид-ион ОН- более эффективно сольватируется водой, чем алкоголят-анион, и остается в водной фазе.
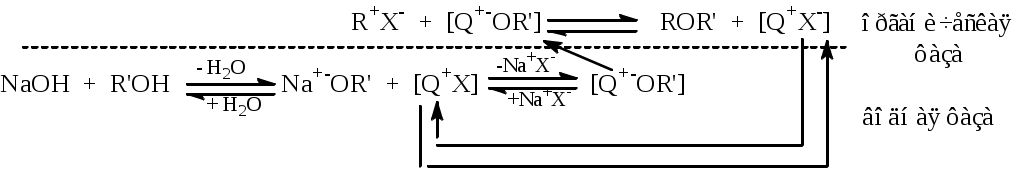
Повышение скорости процесса объясняется повышением нуклеофильности алкоголят-иона,который, во-первых, при переходе из водной фазы в органическую теряет гидратную оболочку, мало сольватирован (в гомогенных условиях десольватация аниона нуклеофила затруднена, особенно в протонных растворителях); во-вторых, его реакционная способность повышается при замене катионов калия или натрия на больший по размеру катион МФ-катализатора.
1) Пример успешного получения простых эфиров в условиях МФК:
2) Пример синтеза диариловых эфиров. Введение в качестве катализатора хлорида гексадецилтриметиламмония (ГДТМАХ) в реакцию п-нитрохлорбензола с п-метоксифенолом в присутствии 25%-го раствора KOH повышает выход эфира с 67% до 98%:
1.5. Замена атома галогена на меркапто- и алкил(арил)тио- группы
- синтез тиоспиртов и тиоэфиров - осуществляется с помощью реагентов, содержащих гидросульфид-, сульфид- и алкил(арил)тио-ионы.
Вместо сульфидов металлов, которые в результате гидролиза выделяют сероводород, можно использовать тиомочевину:
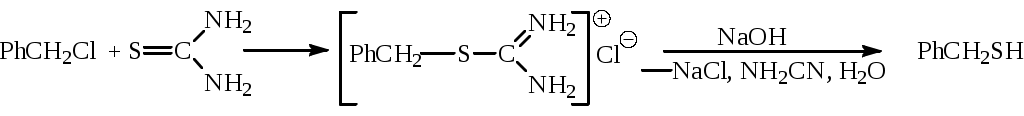
Субстратами являются:
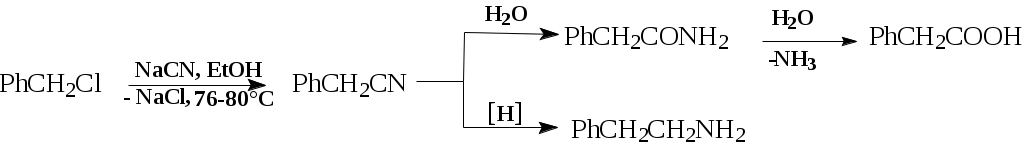
- Алкилгалогениды. Реакцию, обычно, проводят в среде этанола:
- Активированные алкил- и арилгалогениды, которые, обычно, дают хорошие выходы. Под действием сульфид-иона (S-2) можно получить диалкил- и диарилсульфиды:.
- Неактивированные арил- и алкилгалогениды, например, винилгалогениды, которые реагируют с сильным нуклеофилом Alk(Ar)S- анионом в полярных апротонных растворителях (диметилформамид, диметилсульфоксид) или в жидком аммиаке:
Синтез симметричных и смешанных сульфидов в условиях МФК осуществляют с высокими выходами из первичных и вторичных алкилбромидов и меркаптанов или тиофенолов в присутствии водных растворов щелочи. В качестве катализаторов межфазного переноса использовались ониевые соли, краун-эфиры и криптаты. Исследование механизма реакции тиофенола с октилбромидом в щелочной двухфазной системе подтверждает SN2 характер замещения:
1.6. Замена атома галогена на аминогруппы –NH2, -NHR, -NR2
А.Замена галогена в первичных и вторичных алкилгалогенидах на амино группу осуществляется нагреванием их со спиртовым, водным или водно-спиртовым раствором аммиака, первичного или вторичного амина под давлением (в автоклаве). При этом образуется смесь солей первичных, вторичных, третичных аминов и четвертичных солей аммония:

- Третичные алкилгалогениды в этих реакциях обычно не применяют, так как в условиях реакции идет элиминирование с образованием алкенов.
- Температура реакции зависит от активности галогенида и нуклеофила и колеблется в широких пределах (50 – 150оС).
- Выход первичного аминаможно повысить, применяя большой избыток аммиака и добавляя карбонат или хлорид аммония. Однако, даже в этом случае образуется смесь соединений, которые приходится разделять.
Лишь -галогенкарбоновые кислоты при действии большого избытка концентрированного водного раствора аммиака и карбоната аммония при 40-50°С образуют - аминокислоты (первичные амины) с выходом 60-70%.
- Среди наиболееприменяемыевсинтезе БАВ селективных методов получения аминов из галогенидов необходимо отметить:
1. Синтез первичных и вторичных аминов из амидов сульфокислот:
2. Синтез первичных аминов по Габриэлю из фталимида:
 Гидразинолиз идет при нормальном давлении, в то время как гидролиз приходится проводить при высоких температурах под давлением.
Гидразинолиз идет при нормальном давлении, в то время как гидролиз приходится проводить при высоких температурах под давлением.3. Синтез вторичных аминов из азометинов и алкилгалогенидов:

Б. Замена галогена на аминогруппу в неактивированных галогенаренах осуществляется действием раствора аммиака при высокой температуре (