реф. Взаимоотношения аутофагии,апоптоза и некроза
 Скачать 479.36 Kb. Скачать 479.36 Kb.
|

|
Государственное бюджетное образовательное учреждение высшего профессионального образования «Оренбургская государственная медицинская академия Министерства здравоохранения и социального развития Российской Федерации» Кафедра патологической физиологии Реферат Тема: «Взаимоотношения аутофагии,апоптоза и некроза» Выполнил: Студент 21М группы Медико-профилактического факультета 2 курса Губайдуллин Р.Р. Проверила: Доцент, к. мед. н. кафедры патофизиологии Панфилова Т.В. г. Оренбург, 2022 г Содержание: 1.Введение………………………………………………………………..…3 2.Аутофагия………………………………………………………………...4 3.Апоптоз……………………………………………………………………8 4.Некроз……………………………………………………………………11 5.Взаимодействие между путями клеточной смерти……………………15 6.Заключение……………………………………………………………...17 7.Список литературы…………………………………………………......18 1.Введение: Считается, что основной принцип действия иммунной системы заключается в распознавании чужого или измененного своего и его последующем удалении. Классическим примером иммунного распознавания чужого являются реакции врожденного и приобретенного иммунитета против микроорганизмов (бактерий, вирусов). Иммунное распознавание измененного своего ассоциировано с аутоиммунными заболеваниями. С развитием представлений о (за)программированной клеточной смерти (ПКС) стала важной оценка связи иммунитета с поддержанием клеточного гомеостаза в макроорганизме. Всякие изменения клеток в процессе роста и дифференцировки, старения, естественного отмирания, метаболической дисфункции, стресса, воздействия патологического процесса (инфекция, стерильное воспаление) должны рассматриваться иммунной системой как нарушения клеточного гомеостаза. Оценке роли ПКС в запуске иммунных реакций посвящен данный реферат. На основании морфологических и биохимических критериев выделяют три основных типа ПКС: апоптоз (ПКС I типа), аутофагия (ПКС II типа) и некроз (ПКС III типа) ПКС типов I и II имеют определенные генетически механизмы реализации, поэтому называются активными. ПКС III типа (первичный некроз в результате внешнего повреждения) является неуправляемым, поэтому называется пассивным. Дополнительно выделяют вторичный некроз как конечный результат апоптоза, управляемый некроз (некроптоз) и другие пути гибели клеток. Перечень известных типов клеточной смерти регламентирован Номенклатурным комитетом. Характеристика трех основных типов ПКС представлена в таблице. 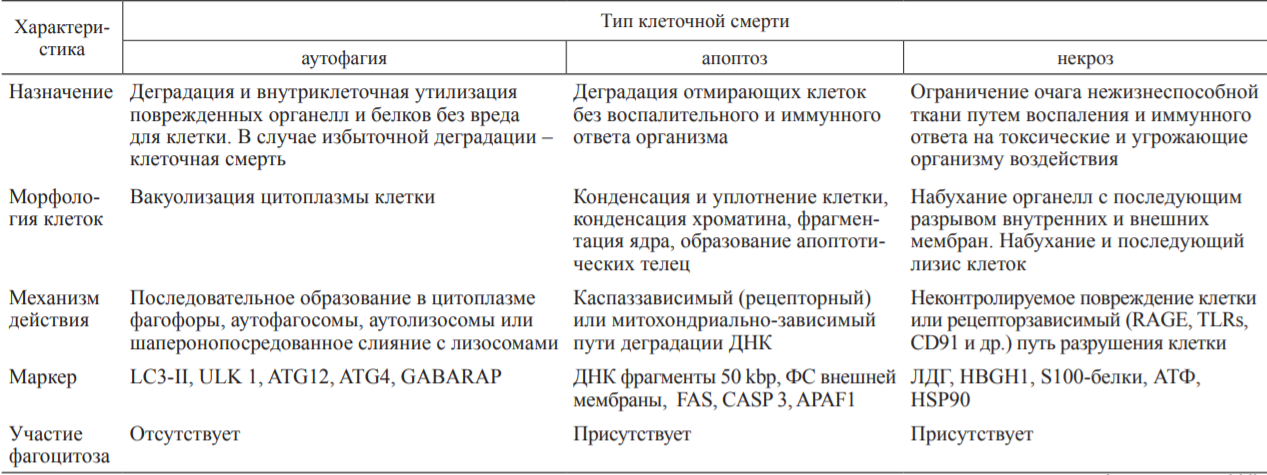 Внимание иммунологов к клеточной смерти определяется тем, что не только инфекционные антигены и молекулярные образы (паттерны) патогенов (pathogen-associated molecular patterns - PAMPs), отличающие его от макроорганизма, но и продукты повреждения собственных клеток (damage-associated molecular patterns - DAMPs) вызывают воспаление и иммунный ответ. P. Matzinger подчеркнула, что для иммунной системы важно распознавание и ответ на сигналы опасности, образующиеся в результате повреждения тканей (клеток), а не выяснение различий между своим и чужим. 2.Аутофагия: Аутофагия - процесс прижизненной утилизации (деградации с помощью лизосом) измененного метаболитами содержимого цитоплазмы для поддержания клеточного и энергетического гомеостаза. Аутофагию рассматривают как преимущественно «запрограммированное выживание клетки». Стресс вызывает аутофагию, а избыточная активность аутофагии ведет к клеточной смерти. Недостаточность аутофагии провоцирует накопление метаболитов, связанных со старением, дегенеративными процессами в нервной ткани и печени, аутоиммунные, легочные заболевания (особенно на фоне курения). Показана связь аутофагии с болезнью Крона, муковисцидозом, ожирением, сепсисом. Основной тип аутофагии - макроаутофагия, включающая этапы инициации, нуклеации, элонгации и слияния (с лизосомой). Измененные белки цитоплазмы (в результате стресса, недостатка энергетического обеспечения), поврежденные митохондрии, избыточный эндоплазматический ретикулум (ЭР), пероксисомы транслоцируются к мембранам органелл благодаря комплексированию с белками ULK 1/2, Atg13, Atg101, fIp-200. На мембранах органелл (ЭР, митохондрии, аппарат Гольджи) эти белки формируют комплекс I, включающий дополнительно белки Vps34, Beclin I. Vps15, Atg14L. Вокруг комплекса I образуется внутренняя мембрана фагофоры. Формирование аутофагосомы (диаметром 0,3-1 мкм) с двойной мембраной требует участия LC3 II.образующегося в результате липолизации фосфатидилэ-таноламином цитозольного белка LC3, и комплекса белков Atg5-Atg12/Atg16L1. Последующее созревание аутофагосомы в аутофаголизосому осуществляется путем слияния с лизосомами с помощью комплекса белков II, включающего Vps34, Beclin 1, UVRAG. В аутофаголизосоме осуществляется деградация измененных белков под действием гидролаз и высвобождение в цитоплазму питательных и энергоемких субстанций. Кроме макроаутофагии выделяют микроаутофагию (когда захват содержимого цитоплазмы осуществляется путем инвагинации мембраны лизо-сом) и шаперонопосредованную аутофагию (когда доставка цитоплазматического материала в лизосомы осуществляется с помощью белков-шаперонов). В связи с наличием в цитоплазме клетки измененных своих и чужеродных макромолекул процесс аутофагии, являясь метаболическим, выступает еще как механизм распознавания и утилизации внутриклеточных микроорганизмов (вирусы, бактерии, простейшие), несущих PAMPs. Проникновение в цитоплазму микроорганизмов и их продуктов запускает механизмы аутофагии в качестве клеточной автономной защитной системы-cell-autonomous defense system. Разделение цитоплазмы клетки на отдельные, ограниченные (эндо)мембранами участки и органеллы (т. е. компартментализация) предполагает наличие в каждом их них своего набора рецепторов, распознающих чужеродные PAMPs и измененные собственные DAMPs. Это создает многоступенчатую систему защиты от патогенов, проникших внутрь клетки. На каждом этапе продвижения патогена в клетке происходит распознавание ДНК, агрегированных собственных белков, комплекса микробов и сывороточных белков. Патоген сталкивается с различными ферментами; NO и H2O2; наличием или недостатком питательных веществ. Микробы активируют рецепторы на эндомембранах цитоплазмы, что ведет к формированию инфламмасомы, продукции интерлейкина (ИЛ)-1р и ИЛ-18. Попадание патогена в аутофаголизосомы резко изменяет условия его существования за счет действия рН, гидролаз, супероксидных анионов. При этом возможны персистенция патогена (длительная для M. tuberculosis, короткая для других бактерий) в аутофагосомах либо разрушение патогена в аутофаголизо-сомах. Toll-like receptors (TLRs) распознают попавшие в цитоплазму макрофагов бактериальный липополисахарид (ЛПС), вирусную однонитчатую рибонуклеиновую кислоту (онРНК), другие полимерные нуклеиновые кислоты. При аутофагии в распознавании внутриклеточных патогенов (Str. pyogenes, M. tuberculosis, BCG, Salmonella, вирусы) участвуют TLRs, RLRs (retinoid acid inducible gene I-like receptors), NLRs (nucleotide oligomerization domain- like receptors). TLR3, распознающий РНК вирусов, локализуется в эндосомах клетки; TLR7, TLR8, TLR9, распознающие РНК и ДНК вирусов и бактерий, CpG-мотивы нуклеиновых кислот микробного происхождения, - в эндолизосомах. RLRs, распознающие РНК вирусов, и NLRs, распознающие PAMPs (мурамил дипептид, токсины, кристаллы солей, другие компоненты) бактерий, вирусов, клеточные продукты химического воздействия и УФ-облучения, расположены в цитоплазме. Важной функций TLRs является обеспечение жесткого контроля за нормальной (комменсальной) микрофлорой кишечника. PAMPs, распознаваемые TLR1, TLR2, TLR4, TLR5, TLR6, вызывают образование в инфламасоме цитокинов воспаления ИЛ-ф и ИЛ-18. PAMPs, распознаваемые TLR7, TLR9, стимулируют продукцию интерферона-а (ИФНа) и ИФНр, что способствует формированию Th1 иммунного ответа. Продукция ИЛ-1Р и ИЛ-18 защищает клетки от вируса гриппа и бактерий рода Shigella соответственно. А вызванный в результате активации инфламмасом пироптоз (гибель клеток с признаками апоптоза и некроза) губителен для сальмонелл, легионелл и других бактерий. Активация TLR4 разрушает связь Bcl-2 c белком Beclin 1, что ведет к образованию фагосомы из фагофоры. Активация TLRs индуцирует быстрый переход Lc3 из цитоплазмы в фагосому, активацию клетки, способствует созреванию фагосомы и слиянию ее с лизосомой. L. monocytogenesis в цитоплазме клетки распознают NLRs и TLR2, а S. flexneri распознают NLRs, что приводит к деградации микробов механизмами аутофагии с участием инфламмасом. При захвате живых бактерий (в отличие от мертвых) в инфицированную клетку попадает микробная мРНК, которая создает дополнительный сигнал опасности (vita-PAMPs), активирующий инфламмасомы типа NLRP3 и TRIF-зависимую продукцию ИФНр. Таким образом, аутофагия выступает как механизм деградации микроорганизмов при их попадании в цитоплазму клетки и распознавании патогенассоциированными рецепторами. Аутофагия участвует в презентации антигенов Т-клеткам. Образование протеосом, ассоциированных с ЭР, или аутофагосом создает благоприятные условия для контакта мембранно-связанных молекул MHC I или II классов с пептидами и последующей передачи их комплексов на внешнюю мембрану антигенпредставляющих клеток для индукции соответственно CD8- или CD4- зависимых Т-клеточных реакций. Белки аутофагии LC3 и GABARAP в аутофагосомах повышают в 20 раз сродство собственных и чужеродных пептидов к молекулам MHC II класса. Блокирование гена аутофагии Atg5 подавляет образование CD4+ Т-клеточного (Th1) ответа на вирус простого герпеса или ВИЧ-1, а также препятствует распознаванию В-клеток, инфицированных вирусом Эпштейна-Барр. Аутофагия в тимическом эпителии является основой негативной селекции аутореактивных Т-клеток. Блок гена аутофагии Atg5 приводит к аутоиммунному CD4+ Т-клеточному пролиферативному заболеванию мышей и накоплению апоптотических CD4+ и CD8+ Т-клеток. Дефицит аутофагии в периферических Т-клетках вызывает ускоренную клеточную смерть наивных, но не Т-клеток памяти, что связывают с продукцией супероксидных анионов при активации наивных Т-клеток. Важной функцией аутофагии является изоляция поврежденных митохондрий, генерирующих супероксидные анионы, как источник стресса и повреждения (вплоть до гибели) самой клетки. Аутоиммунный ответ при сахарном диабете и аутоиммунном гепатите вызывают аутоантигены GAD65 (глутамат декарбоксилаза 65) и SMA (мутантная к-легкая цепь иммуноглобулинов), которые подвергаются в цитоплазме шаперо-нопосредованной аутофагии с участием HSC70 и связанного с лизосомами мембранного белка LAMP-2A соответственно. После деградации в лизосомах они вместе с молекулам MHC II класса презентируются аутореактивным cD4+ Т-клеткам. Образование в аутофаголизосомах цитрулированных пептидов под действием пептидиларгинин деаминаз и формирование их комплексов с молекулами MHc II класса является основой аутоиммунного cD4+ Т-клеточного ответа при ревматоидном артрите - РА. В Т-клетках мышей линии MRL с лимфопролиферативным синдромом, аналогом системной красной волчанки (СКВ) человека, выявляется значительное количество аутофагосом в Т-клетках, что объясняют их длительным выживанием. Продукция супероксидных анионов митохондриями макрофагов способствует переваривание бактерий в процессе аутофагии. Бактерии, распознаваемые NLRs, стимулируют аутофагию в фибробластах. В дендритных клетках (ДК) это приводит к представлению пептидов бактерий вместе с молекулами MHC II класса CD4+ T-клеткам. Важной защитной функцией аутофагии является способность снижать уровень собственных DAMPs в цитоплазме и сдерживать секрецию ИЛ-ф и ИЛ-18 в ответ на экзогенные источники DAMPs. Механизмы аутофагии обеспечивают деградацию инфламмасом - комплекса белков, превращающих прокаспазу-1 в каспазу-1, конвертирующую про- ИЛ-ф и про-ИЛ-18 в секретируемые активные цитокины. Блокировка гена аутофагии Atg16L1 приводит у мышей к повышенной продукции ИЛ-ф и ИЛ-18, воспалению, повышению уровня смертности при антигенной стимуляции декстран сульфатом. Внеклеточные цитокины влияют на процессы аутофагии бактерий и их переваривание в фаголизосомах. Цитокины ТЫ-зависимого ответа ИФНу и фактор некроза опухолей а (ФНОа) стимулируют аутофагию. Цитокины №2-зависимого ответа ИЛ-4 и ИЛ-13, наоборот, снижают образование фаго-лизосом и повышают внутриклеточное выживание M. tuberculosis. Дифференцировка Т-клеток в Th1 и Th2 in vitro характеризуется большим и меньшим образованием ауто-фагосом соответственно. Внутриклеточные инфекционные агенты (цитомегаловирус, ВИЧ, вирус герпеса простого I, вирус гриппа А, йерсинии, листерии, шигеллы, сальмонеллы, E. coli и др.) избегают иммунного ответа путем ослабления процесса аутофагии. Аутофагия является физиологическим процессом самообновления клетки, которое при стрессовых воздействиях может привести к ее гибели. В то же время естественное отмирание клеток (у человека от 50 до 500 млрд клеток ежедневно) осуществляется преимущественно путем апоптоза. 3.Апоптоз: Апоптоз обеспечивает удаление отмирающих клеток посредством фагоцитоза без воспаления, губительного для макроорганизма, или сопровождает очаг воспаления для его ограничения и окончательного заживления. Формирование иммунной системы и созревание антигенспецифических Т- и В-лимфоцитов также сопровождается массовым апоптозом клеток. Апоптоз обеспечивает поддержание клеточного гомеостаза, стимуляцию клеточной регенерации, заживление ран. Апоптотические клетки (АК) утилизируются соседними клетками эпителия, эндотелия, фибробластами, макрофагами, ДК. При заболеваниях и переливании хранившейся донорской крови в периферической крови, лимфоузлах, костном мозге выявляются апоптотические тельца диаметром 0,2 мкм, образующиеся из АК. Выделяемые АК липидные медиаторы (лизофосфа-тидилхолин, сфингозин-1-фосфат), рибосомальный dRP S19, EMAP II эндотелиальных клеток, TyrRS синтетазу, тромбоспондин 1, растворимый рецептор к ИЛ-6, фракталкин (CX3-CR1L), нуклеотиды АТФ и УТФ привлекают фагоциты. При этом лактоферрин, выделяемый клетками слизистых и нейтрофилов при апоптозе, избирательно подавляет хемотаксис нейтрофилов, но не макрофагов. Поверхностная экспрессия фосфатидилсерина (ФС), других окисленных липидов и калретикулина является признаком ранних АК, распознаваемых рецепторами макрофагов (стабилин-2, CR3, рецепторы-мусорщики (scavenger receptors), CD91, CD31, TIM4, CD36, steroid receptor activator 1; TAM-рецепторы (Ty-ro2, Ax1, Mer); LRP-1). Молекулярные маркеры АК получили общее название apoptotic cell-associated molecular patterns (ACAMPs). Макрофаги распознают апоптотические клетки посредством нескольких апоптозассоциированных рецепторов одновременно для быстрого удаления клеток на ранних этапах апоптоза. Экспрессия поверхностного CD31 (и/или CD47) на АК предотвращает их захват макрофагами. Важно, что рецепторы макрофагов, распознающие АК и апоптотические тельца, отличаются от рецепторов, распознающих PAMPs и DAMPs. Более того, активация рецепторов, различающих АК и апоптотические тельца, способствует подавлению распознавания макрофагами PAM-Ps инфекционных агентов через TLRs. Распознавание АК и апоптотических телец облегчается участием сывороточных опсонинов Gas6, MFG-E8, P2GP1, аннексина I, С-реактивного белка (СРБ), пентраксина PTX-3, коллектинов, dq-компонента комплемента, сурфактантов SP-A и SP-D (в легочной ткани) и т. д.. При этом опсо-нин MFG-E8, участвующий в захвате АК макрофагами, одновременно подавляет фагоцитоз некротических клеток (НК) и их иммуногенность для ДК. C1q взаимодействует с ФС ранних АК, а коллектин маннозасвязывающий лектин (MBL) - с поздними АК. Калретикулин (в комплексе с CD91), пентрак-сины СРБ, SAP (компонент сывороточного амилоида Р); фи-колины взаимодействуют с поздними АК. Оценивая роль системы комплемента и естественных антител в клиренсе АК. Ряд авторов определили, что лизофосфатидилхо-лин, появляющийся (и частично секретируемый) на поверхности АК, является мишенью естественных антител - IgM, а также маннозасвязывающих белков, других коллектинов. Их взаимодействие в свою очередь приводит к связыванию с C1q, C3b/bi. В результате АК фагоцитируются без активации выброса макрофагами провоспалительных цитокинов. Аутоиммунные реакции с участием антикардиолипиновых антител класса G, наоборот, протекают с участием комплемента и аутоантител к фосфолипидам мембран поздних АК. Важно, что апоптотические тельца на ранних этапах апоптоза покрыты элементами ФС-содержащей внешней мембраны клеток, а на поздних этапах - элементами эндоплазматических мембран. И если антигенная презентация ранних апоптотических телец вызывает образование иммунорегуляторных Т-клеток (Treg), то контакт поздних апопто-тических телец с ДК вызывает образование ТЫ7-клеток. Апоптотические нейтрофилы (и внешние мембраны лизированных нейтрофилов) вызывают продукцию трансформирующего ростового фактора в (ТРФр) макрофагами, а внутреннее содержимое лизированных нейтрофилов - образование ИЛ-8, ФНОа, хемокина MIP-2. В очаге воспаления сами нейтрофилы проявляют «каннибализм», фагоцитируя апоптотические нейтрофилы (например, индуцированные УФ-облучением). Этому способствуют дополнительная активация TLRs эффекторных нейтрофилов и цитокины ФНОа и гранулоцитарно-макрофагальный колониестимулирующий фактор (ГМ-КСФ), но не ИЛ-1-р, ИЛ-6, ИЛ-8, ИЛ-12, ИЛ-17. В очаге воспаления макрофаги являются основными фагоцитами АК. Это не приводит к продукции провоспалительных цитокинов (ИЛ-1р, ФНОа, ИЛ-6, ИЛ-12), но вызывает образование иммуносупрессорных ИЛ-10, ТРФр, простагландина Е2 (ПГЕ2). Формируется иммунная толерантность к антигенам АК и одновременно к другим антигенам, включая PAMPs микроорганизмов, которая опосредуется СЭ8а+ДК. ДК, стимулированные АК, представляют антиген(ы) только CD8+ Т-клеткам, а ДК, стимулированные НК, представляют антиген(ы) CD4+ и CD8+ Т-клеткам . Иммуносупрессия, развивающаяся в результате массового образования АК и их захвата макрофагами, лежит в основе лечебного действия экстракорпорального фотофереза у пациентов с хроническими воспалительными заболеваниями. Длительно протекающий процесс апоптоза в очаге воспаления может привести к формированию фиброза, что связывают со способностью макрофагов, фагоцитировавших АК, секретировать ТРФр и другие ростовые факторы. В то же время подавление воспаления, усиление репаративных процессов при фагоцитозе АК приводит при наличии генетической предрасположенности к аутоиммунным заболеваниям (СКВ, хроническое обструктивное заболевание легких). В норме В1-подобные клетки с фенотипом CD43+CD27-IgM+ или cD24++cD38++cD27- IgM+ являются основным источником естественных антител к поверхностным молекулам АК. Значительное количество АК в герминативных центрах лимфоузлов у пациентов с СКВ обеспечивает длительное выживание и костимуляцию аутореактивных В-клеток, активированных однониточной ДНК, нуклеосомами, другими клеточными антигенами. Это связано с Oq-зависимым генетическим дефектом быстрого клиренса ранних АК и накоплением поздних АК с признаками вторичного некроза. Образующиеся низкоаффинные антитела класса IgM взаимодействуют с клетками, находящимися на ранних стадиях апоптоза, а высокоаффинные антитела класса IgG - с клетками, находящимися на поздних стадиях апоптоза. Плазмацитоидные ДК и активация ДНК-связывающих TLR9 В-клеток обеспечивает Т-независимое образовании аутоантител. Индуцируемая АК продукция иммуносупрессорного ИЛ-10 значительно снижена при стимуляции В-клеток иммунными комплексами, включающими хроматин, или апоптотическими тельцами, образующимися на поздних этапах апоптоза. Элиминация АК осуществляется в основном на ранних этапах апоптоза, когда экспрессия на внешней мембране ФС и калретикулина сигнализирует об «измененном своем». Ранние этапы апоптоза обратимы, их продление обеспечивает фагоцитоз большинства АК и формирование толерантности иммунной системы. Переход клеток на поздние этапы апоптоза характеризуется снижением уровня гликозилирования поверхностных молекул, фрагментаций ядерной ДНК и признаками вторичного некроза, вызывающего воспаление и иммунный ответ. Основными путями запуска апоптоза клеток являются рецепторный (extrinsic), обусловленный внешним воздействием, или стрессиндуцированный (intrinsic), связанный с внутренним воздействием. Рецепторный путь запуска апоптоза клетки опосредован рецепторами смерти (death receptors), включающими Fas, TNFR (рецептор I типа к ФНОа), TRAIL, Apo2/Apo3. Активация каспаз является ключевой для апоптоза и последовательность их включения достаточно описана в литературе. Стрессиндуцированный (митохондриальный) путь апоптоза связан с высвобождением цитохрома С из митохондрий и регулируется белками семейства Bcl2. Каспаззависимая активация и повышение уровня супероксидных анионов (преимущественно за счет повреждения митохондрий) определяют иммуносупрессорное действие АК. Толерогенное действие АК, считается, опосредуется Heg-клетками, вызывающими TRAIL-индуцированную гибель CD4+ Т-клеток-хелперов. Оба пути апоптоза приводят к поверхностной экспрессии ФС, фрагментации ДНК ядра, образованию апоптотических телец и их быстрому фагоцитозу. Это предотвращает иммунный ответ на отмирающую клетку, продукцию макрофагами цитокинов воспаления, презентацию ДК клеточных антигенов. При инфицировании клетки проявляют признаки раннего апоптоза (экспрессия на клеточных мембранах ФС, начало фрагментации ДНК) и NF-кБ-зависимого пути клеточной активации. Одновременно клетки сдерживают репликацию патогенов без образования DAMPs, свойственных некротическим клеткам. Дефекты звеньев апоптоза (преимущественно митохондриальнозависимого пути активации), или запоздалый запуск апоптоза приводят к распространению инфекции (вызванной Legionella pneumonia, Pseudomonas aeroginosa, Helicobacter pylori), сепсису. Многие вирусы содержат ингибиторы каспаз, а Chlamydiae и Coxiella burnetii блокируют выход цитохрома c из митохондрий и апоптоз клетки, что обеспечивает жизненный цикл патогена на ранних этапах инфекции. Захват АК, содержащих бактерии, вызывает созревание ДК, воспаление, полноценный (Th17) иммунный ответ, при захвате неинфицированных АК признаки созревания ДК и воспаления отсутствуют, формируется иммуносупрессия. Стратегия ограниченной репликации патогена в АК выгодна отсутствием сильного иммунного ответа на некроз клеток и массовый выброс бактерий во внеклеточное пространство. 4.Некроз: Клетки, отмирающие в результате травмы, дегенеративных процессов, воздействия патогена, эффективно утилизируются путем некроза. Некроз демаркирует нежизнеспособную ткань, подлежащую уничтожению и последующему восстановлению. Некроз клеток всегда сопровождается воспалением и ведет к выраженному иммунному ответу и последующей репарации тканей. НК характеризуются разрушением внешней клеточной мембраны и поступлением во внеклеточное пространство скрытых внутриклеточных молекул (см. таблицу), что вызывает токсическую реакцию окружающих здоровых клеток и иммунный ответ. Первичный некроз клеток не зависит от действия каспаз и является прямым результатом внешнего травматического повреждения или запрограммированных генетически событий, связанных с повреждением белка митохондриального матрикса циклофилина Д; воздействием на рецепторы смерти или TLR3/TLR4 и рецепторнезависимым повреждением ДНК. Оксидативный стресс клетки, активные формы кислорода являются индукторами (управляемого) некроза. Вторичный некроз - это конечный результат позднего апоптоза, часто он лежит в основе аутоиммунной патологии (СКВ и другие). НК фагоцитируются путем макропиноцитоза после исчезновения поверхностных CD31- и СЭ47-молекул, блокирующих фагоцитоз. НК в отличие от АК вызывают созревание ДК и (Th1) иммунный ответ. НК выделяют внутриклеточные молекулы, провоцирующие воспаление и иммунную реакцию, поэтому они названы аларминами (alarmins) или DAMPs. Они привлекают нейтрофилы в очаг некроза. НК выделяют белки теплового шока (HSP70, HSP90, gp96), калгра-нулины, цитокины (ИЛ-1а, ИЛ-6), формилпептиды митохондрий, РНК, двунитчатую (геномную) ДНК, другие молекулы. Выделение ядерного белка HMGB1 (high-mobility group box 1), связанного в норме с хроматином, является основным маркером (первичного) некроза клеток. При апоптозе и вторичном некрозе HMGB1 удерживается в ядре или находится в цитоплазме или внеклеточно в неактивном (окисленном) состоянии в результате действия супероксидных анионов. Сам HMGB1 является митогеном и хемоаттрактантом, но образуемые им комплексы с однонитчатой ДНК, ЛПС бактерий, нуклеосомой вызывают секрецию макрофагами цитокинов воспаления ФНОа, ИЛ-1р, ИЛ-6, хемокины ИЛ-8, MIP-1a, MIP-ip. Высокий уровень HMGB1 в крови связан с массивным некрозом клеток организма и является маркером системного воспаления. HMGB1 - мощный адъювант образования высокоаффинных антител и созревания ДК. Циркулирующий в кровотоке неокисленный (активный) HMGB1 взаимодействует c TLR2, TLR4, TLR9 и RAGE (receptor for advanced glycation end-products) фагоцитов, вызывая воспалительный ответ. Одновременно HMGB1 (а также HSPs) взаимодействует с CD24 и Siglec-10 на поверхности фагоцитов, что ограничивает воспаление, вызванное DAMPs, но не PAMPs. Разграничение иммунного ответа на патогенассоциированные PAMPs и связанные с повреждением собственных клеток DAMPs происходит на уровне рецепторов клеток. Типичным рецептором для DAMPs является RAGE на клетках иммунной и нервной систем, эндотелиальных клетках, кардиомиоцитах. RAGE распознает белки и липиды, модифицированные в результате неферментативного гликозили-рования и появляющиеся при хронических воспалительных заболеваниях как результат окислительного стресса. RAGE распознает такие продукты НК, как HMGB1 и калгранулины (белки семейства S 100). НК выделяют нуклеиновые кислоты. При этом РНК становится двунитчатой, взаимодействует с TLR3 на ДК, а двунитчатая ДНК - с TLR9 фагоцитов, что приводит к продукции ИФНу, CXCL10 (IP-10), ИЛ-1Р, экспрессии костимулирую-щих молекул (cD40, cD54, cD69, MHc II класса) на поверхности макрофагов и ДК. Для того чтобы не вызвать воспаления, молекулы ДНК подвергаются ферментативному расщеплению, например, каспаз при апоптозе. Дефект ДНКаз, разрезающих двунитчатую ДНК, вызывает у мышей аутоиммунные заболевания (СКВ, полиартрит). Нуклеотиды АТФ и УТФ, в норме находящиеся в цитоплазме, при некрозе клеток выделяются во внеклеточное пространство. Действуя на пуринэргические рецепторы ДК, они вызывают хемотаксис незрелых ДК, образование NALP3 инфламмасом и секрецию ИЛ-1р, Th2 иммунного ответа. Действие АТФ на аллергенактивированные миелоидные ДК провоцирует развитие легочной аллергии и поддержание бронхиальной астмы. Ядерные рибонуклеопротеины (их короткие фрагменты) выделяются при разрушении НК и выступают в качестве DAMPs, стимулируя образование цитокинов и а-хемокинов. Соли ураты, образуемые из мочевой кислоты при разрушении в цитоплазме эндогенной ядерной или микробной ДНК и ионов натрия внеклеточного пространства, стимулируют образование инфламмасом в макрофагах и ДК, синтез цитокинов ИЛ-1Р, ИЛ-18, ИЛ-33, нейтрофильную инфильтрацию, созревание ДК, усиление антигенспецифического Т-клеточного ответа. Стрессиндуцированные цитоплазматические белки-шапероны HSP70, HSP90 при некрозе (но не апоптозе) клеток поступают во межклеточное пространство. Внеклеточные HSP70, HSP90 стимулируют образование цитокинов воспаления (ФНОа, ИЛ-1Р, ИЛ-6, ИЛ-12). Антигенспецифический иммунный ответ на комплекс пептид-HSP значительно возрастает. Клеточными рецепторами HSPs выступают cD91, CD40, TLR2/TLR4/CD14, рецепторы-мусорщики, LOX-1. НК выделяют калгранулины (S100 белки), которые распознаются RAGE рецепторами клеток эндотелия, микроглии, моноцитов и становятся маркерами воспаления (при пневмониях, полиартритах и т. д.). Выделение цитокинов (ИЛ-1, ИЛ-6, ИЛ-33) также может быть результатом стрессового воздействия на клетки и их некротической гибели. Высвобождаемые из НК протеазы и биологически активные молекулы воздействуют на окружающие ткани и отщепляют от них низкомолекулярные фрагменты (гиалуроновая кислота, фибриллярный белок, коллаген, гепарансульфат), которые также вызывают воспаление. Как и при утилизации АК, сывороточные факторы (кол-лектин MBL) связываются с НК, усиливая их распознавание и связывание с калретикулином на поверхности макрофагов. Макрофаги распознают некротические клетки посредством TLRs, лектин С-типа рецепторов Clec9A, RAGE; CD14, CD91, CD40, Mincle (взаимодействующим с SAP-130) и других. Важно, что рецепторы фагоцитов, распознающие НК, не распознают АК и (частично) распознают молекулы (PAMPs) патогенов (микобактерии, грибки и др.). Регулируемый некроз (некроптоз) клеток связан с активностью киназ RIPK1 и RIPK3, проявляется быстрым повышением проницаемости клеточных мембран и высвобождением во внеклеточное пространство внутриклеточных DAMPs. Некроптоз клеток кожи, слизистых, лейкоцитов при ишемической реперфузии вызывает сильный воспалительный ответ. Одновременно он выступает в качестве защитного механизма при вирусной инфекции (при наличии вирусных ингибиторов каспазы 8), а также участвует в поддержании гомеостаза Т-лимфоцитов. Некроптоз инфицированной клетки означает резкое изменение среды обитания внутриклеточных патогенов, что губительно для них. Пироптоз клеток, имея черты апоптоза и некроза, характеризуется образованием инфламмасом как комплекса активированных каспаз и продуцентов цитокинов воспаления ИЛ-1Р и ИЛ-18. Пироптоз эффективно защищает клетки от S. aureus, S. ty-phimurium, P. aeruginosa, L. pneumophila, F.tularensis, B. an-thracis. При этом разные типы специализированных инфлам-масом образуются в ответ на живые бактерии, их токсины, ЛПС, споры, флагеллин, ДНК, РНК вирусов и бактерий. Некроз клеток характеризует продвинутые (не ранние) этапы инфекционного процесса, когда патогены (Shigella, Salmonella, Yersinia, M.tuberculosis) переходят от тактики выживания в апоптотических клетках к тактике разрушения клетки и межклеточного распространения. 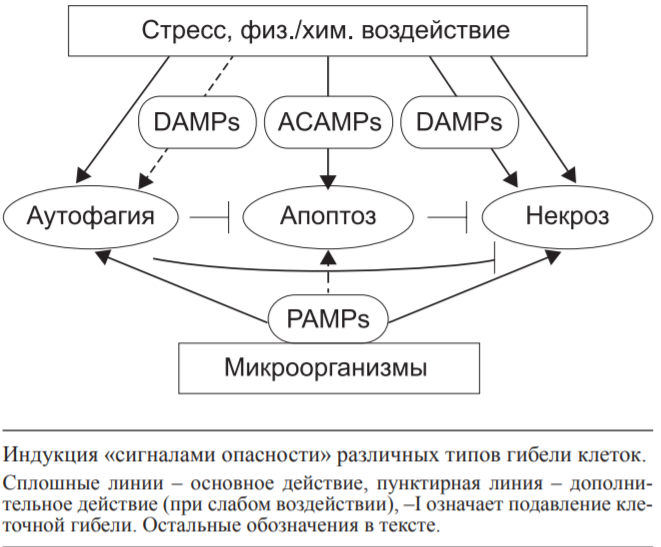 Вторичный некроз как исход апоптоза клеток характеризуется выделением DAMPs нуклеосом (фрагментов геномной ДНК размером 180 пар оснований), HMGB1. Иммуностимулирующее действие таких DAMPs связано с образованием комплексов нуклеосом с HMGB1, характерных для пациентов с СКВ [6, 56]. Вторичный некроз сопровождается массовым высвобождением модицифированных (в результате ферментативной обработки, окисления) аутоантигенов, которые в комплексе с HSPs (и другими DAMPs) вызывают антигенспецифический иммунный ответ. Но только наличие генетической предрасположенности приводит к формированию аутоиммунной патологии 5.Взаимодействие между путями клеточной смерти: Аутофагия и апоптоз клеток рассматривают как механизмы поддержания жизнеспособности многоклеточного организма, а формирование инфламмасом и некрозиндуцированное воспаление считают механизмами ограниченного отмирания тканей для сохранения макроорганизма. Распознавание DAMPs при аутофагии создает дополнительную страховку клеткам макроорганизма в защите от патогенов с неизвестными PAMPs. В результате инфицирования макрофагов L. pneumophila активация инфламмасом вызывает пироптоз и ау-тофагию, которая защищает клетку от пироптоза и патогена. Но недостаточность аутофагии для противодействия патогену ведет инфицированную клетку к пироптозу. Запуск PIRK1-3-зависимого механизма некроптоза предполагает первоначально высокий уровень аутофагии поврежденных митохондрий и при ее неэффективности последующую деградацию клетки. Аутофагия выступает в качестве механизма утилизации фагоцитированных апоптотические телец макрофагами и ДК. Повышение при некрозе клеток уровня HMGBT цитоплазме стимулирует вместе с HSP27 аутофагию (митофагию) митохондрий и подавляет апоптоз. Другие DAMPs (АТФ, белки S100/ калгранулины, двунитчатая ДНК), взаимодействуя с TLRs, также стимулируют аутофагию в очагах апоптоза. Известно, что основной Beclin 1 -зависимый путь аутофагии (макроаутофагия) может быть подавлен анти-апоптотическими белками семейства Bcl-2 и образованием NLRP3-инфламмасом, т. е. возрастание резистентности клетки к апоптотической гибели повышает ее устойчивость к избыточной аутофагии, приводящей к гибели клеток. При фагоцитозе клеток, умерших путем аутофагии или апоптоза, воспаление отсутствует. Блокирование аутофагии в клетке приводит к накоплению в цитоплазме поврежденных митохондрий, супероксидных анионов, активации NALP3-инфламмасомы, воспалению. Взаимодействие DAMPs с рецепторами RAGE стимулирует аутофагию и подавляет апоптоз клеток . При недостаточном выделении DAMPs из НК в очаге повреждения апоптотические клетки вызывают состояние толерантности и снижение воспаления. Созревание ДК вызывают DAMPs из НК, но не ACAMPs из АК. Макрофаги, фагоцитировавшие АК, выделяют ТРФр, что вызывает образование Teg-клеток. При фагоцитозе АК, инфицированных E. coli, макрофаги выделяют ТРФр и ИЛ-6, что ведет к образованию ТЫ7-клеток, а при фагоцитозе НК -Th1 иммунного ответа. При совместном воздействии PAMPs и DAMPs последние выполняют роль адъюванта. Известно, что в зависимости от дозы воздействия (например, ФНОа) клетка погибает путем апоптоза (при низких концентрациях) или некроза (при высоких концентрациях). Связь между апоптозом и некрозом клеток определяется также наличием промежуточных подтипов клеточной смерти - не-кроптоза и других. Разные типы клеточной смерти как результат ответа клеток на внешние (включая микроорганизмы) и внутренние воздействия могут протекать одновременно и регулировать друг друга . До конца неясны механизмы, определяющие выбор пути клеточной смерти, но чем сильнее воздействие, тем сильнее ответ в виде некроза клеток, мощной воспалительной и иммунной реакции макроорганизма. Слабые воздействия (за счет аутологичных apoptotic cell-associated molecular patterns (AcAMPs) или DAMPs, PAMPs нормальной микрофлоры) вызывают интенсификацию аутофагии и апоптоза клеток без очевидной воспалительной и иммунной реакций. 6.Заключение: Гибель клеток макроорганизма (человек, животные), обусловленная внешними или внутренними причинами, вызывает иммунный ответ на повреждение. При этом микробные воздействия всегда дозированы концентрацией и жизнеспособностью патогена, его растворимыми продуктами, локализацией очага повреждения. Сочетанное действие PAMPs и DAMPs, наиболее часто встречающееся в реальных условиях, а также влияние толерогенных апопто-тических клеток на их взаимодействие требуют дальнейшего изучения и оценки иммунологических последствий 7.Список литературы: 1. Ярилин А.А. Апоптоз. Природа феномена и его роль в целостности организма. Патологическая физиология. 1998; 2: 38-48. 2. Green D.R. The end and after: how dying cells impact the living organism. Immunity. 2011; 35 ( 4): 441-5. 3. Бра М., Квинан Б., Сузин С.А. Митохондрии в программированной гибели клетки: различные механизмы гибели. Биохимия. 2005; 70 (2): 284-93. 4. Черников В.П., Белоусова Т.А., Кактурский Л.В. Морфологические и биохимические критерии клеточные гибели. Архив патологии. 2010; 72 (3): 48-54. 5. Galluzzi L., Vitale I., Abrams J.M., Alnemri E.S., Baehrecke E.H., Blagosklonny M.V et al. Molecular definition of cellular death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Different. 2012; 19 (1): 107-20. |