1 часть. 1 спирт этиловый
 Скачать 444.3 Kb. Скачать 444.3 Kb.
|

|
1 вопрос. Идентификация орг ЛВ по функц. группам: 1)спирт этиловый. 1.Проба Лукаса: при реакции вторич или третич спирта при ком t с р-ом Лукаса (смесь конц HCl и ZnCl2) происх образование галогенопроизв, кот распознаётся по образованию характер масла, образующего отдельную от исходного спирта фазу.
2.Иодоформная проба: C2H5OH + 4I2 + 6NaOH → CHI3↓ +HCOONa + 5NaI + 5H2O (жёлт осадок иодоформа со специф запахом) 3. Р-ия образования уксусно-этилового эфира (уксусно-этиловый эфир с характер запахом) 2CH3COONa + 2C2H5OH + H2SO4 → 2СН3СОО-С2Н5 + Na2SO4 + 2H2O 4.Р-ия образ-ия этилбензоата. При взаимодействии этилового спирта с бензоилхлоридом образ этилбензоат с характерным запахом: 5.Р-ия обр-ия ацетальдегида. Этиловый спирт окисл дихроматом калия, KMnO4 и некот др окислителями до ацетальдегида: ЗС2Н5ОН + К2Cr2O7, + 4H2SO4 ---> 3СН3СНО + Cr2(SO4)3 + K2SO4 + 7H2O 2)сложноэфирная группировка. 1. Р-ии гидролиза. Препараты, содержащие в своей структуре сложноэфирную группу, подвергаются щелочному или кислотному гидролизу. 1.1. Р-ии щелоч гидролиза. Под действием щелочей образ соль орг к-ты и спирт (запах йоформа) 1.2. Р-ии кислот гидролиза.Под действием кислоты образ орг кислоты (появл розовое окр-ие) 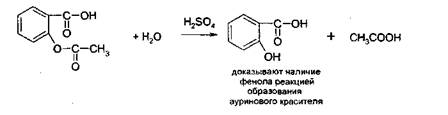 2.Гидроксамовая реакция. При взаим с гидроксиламином образ гидроксамовые к-ты, кот после подкисления HCl образ окраш гидроксаматы с солями железа (III) или меди (II)→вишневое окр-ие 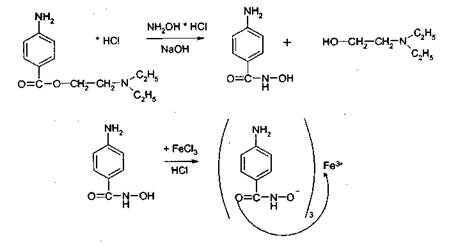 2 вопрос. Идентификация орг. ЛВ по функц. группам: 1). Альдегидная группа: -Р-ия с р-ом Фелинга – «р-ия мед зеркала» ↓кирпично-крас -Р-ия с р-ом Толленса – «р-ия серебр зеркала» 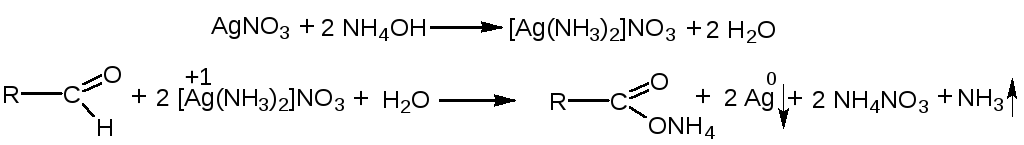 -Р-ия с р-ом Несслера – осадок черн цв -Р-ия с гидроксиламином 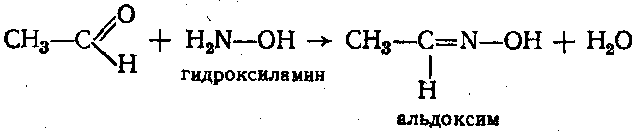 -Р-ия с фенилгидразином 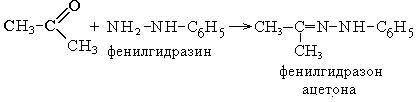 2). Первич аромат аминогруппа - 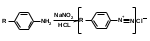 Образование азокрасителя: Образование азокрасителя:1-я стадия – получение соли диазония: 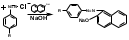 2-я стадия – образование азокрасителя: Стрептоцид – оранж↓; сульфацил-натрий – оранж-крас↓; сульфален – вишнево-крас окр-ие, затем оранж-крас↓ - 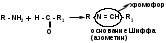 Р-ия конденсации с альдегидами Р-ия конденсации с альдегидамиВанилин-желтое, фурацилин-красное или малиновое, формальлдегид-желт-оранж 3 вопрос. Идентификация орг. ЛВ по функц. группам: 1). первич алифатическая группа Нингидриновая проба. Первич алиф амины окисл нингидрином при нагревании (исп аминок-ты, до сине-фиол окр-ия) 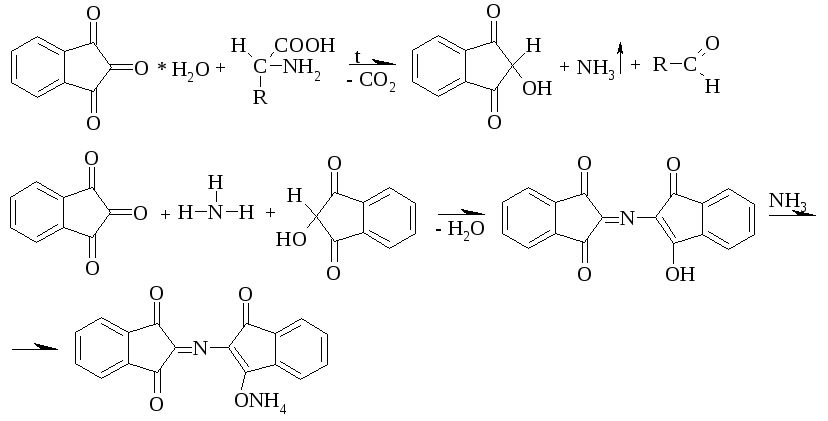 2). салицилат-ион -С хлоридом железа(III) – сине-фиол или красно-фиол окр-ие 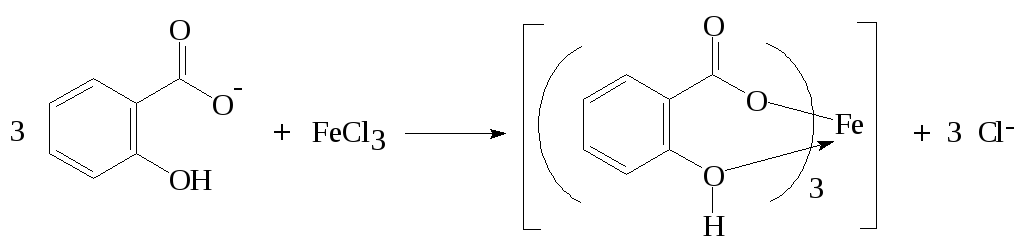 -С сульфатом меди(II) - зел цв 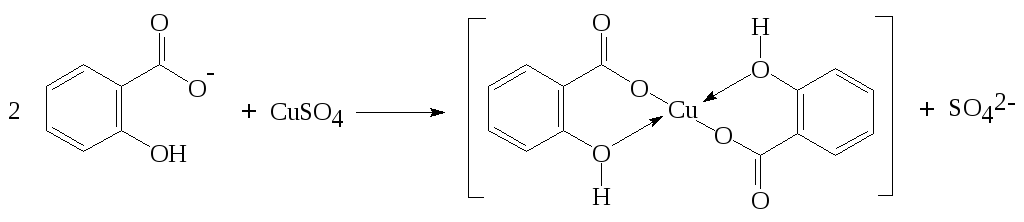 4 вопрос. Идентификация орг ЛВ по функц группам: 1) сложноэфирная группа -Кислотный гидролиз 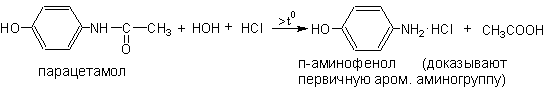 -Щелочной гидролиз 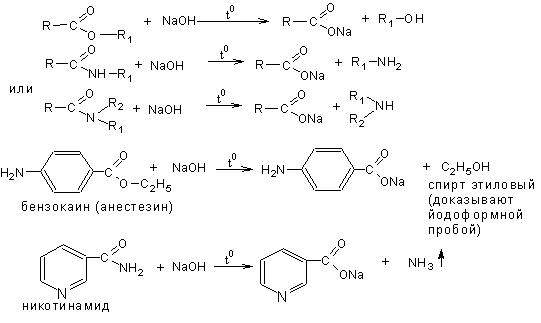 2)вторич аромат аминогруппа (пиперазин→бел пластинчатые кристаллы; резерпин→флуоресц р-ры) 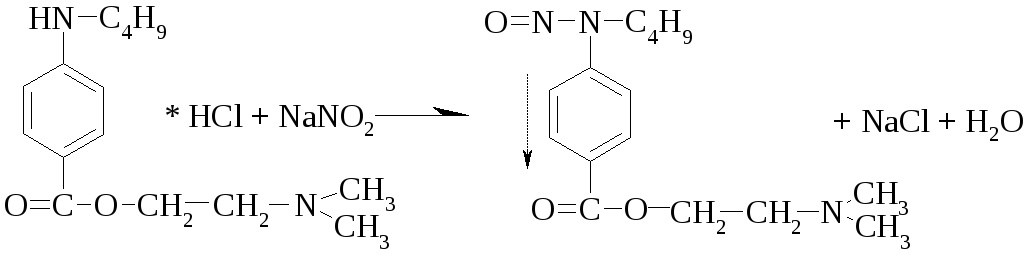 5 вопрос. Идентификация орг ЛВ по функц группам: 1) гидразидная группа -Р-ия серебр зеркала (с р-ом Толленса) 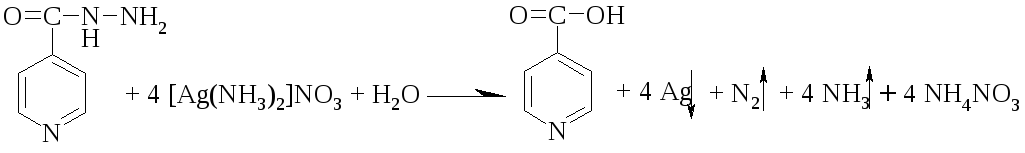 2)альдегидная группа -Р-ия с р-ом Фелинга – «р-ия мед зеркала» ↓кирпично-крас -Р-ия с р-ом Толленса – «р-ия серебр зеркала» -Р-ия с р-ом Несслера – осадок черн цв 6 вопрос. Идентификация орг ЛВ по функц группам: 1) карбоксильная группа -р-ия этерификации 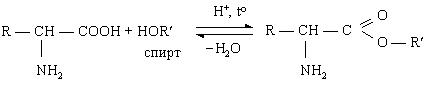 2)органически связанный фтор -минерализации фторотана расплавленным металл натрием CF3 –CHClBr → 3 NaF + NaBr + NaCl + … 7 вопрос. Идентификация орг ЛВ по функц группам: 1) спиртовый гидроксил -р-ия этерификации  -р-ия комплексообр-ия  2)фенольный гидроксил -р-ия бромирования  -р-ия азосочетания  8 вопрос. Идентификация ЛВ по функц группам: 1) пиридиновый цикл  2 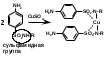 ) сульфамидная группа ) сульфамидная группа 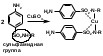 (стрептоцид – зел-голубой; СУЛЬФАЦЕТАМИД-НАТРИЙ сульфацил-натрий – голубовато - зел, не изменяющийся при стоянии; сульфален – серо-зеленый) (стрептоцид – зел-голубой; СУЛЬФАЦЕТАМИД-НАТРИЙ сульфацил-натрий – голубовато - зел, не изменяющийся при стоянии; сульфален – серо-зеленый)9 вопрос. Идентификация ЛВ по функц группам: 1) альдегидная группа -Р-ия с р-ом Фелинга – «р-ия мед зеркала» ↓кирпично-крас -Р-ия с р-ом Толленса – «р-ия серебр зеркала» -Р-ия с р-ом Несслера – осадок черн цв 2) первич аромат аминогруппа - Образование азокрасителя:1-я стадия – получение соли диазония 2-я стадия – образование азокрасителя: 10 вопрос. Идентификация ЛВ по функц группам: 1 ) сульфамидная группа (стрептоцид – зел-голубой; СУЛЬФАЦЕТАМИД-НАТРИЙ сульфацил-натрий – голубовато - зел, не изменяющийся при стоянии; сульфален – серо-зеленый)2)простая эфирная группа -р-ия кислотного гидролиза (опред t плавления образовавшегося бензгидрола- 62-67гр) 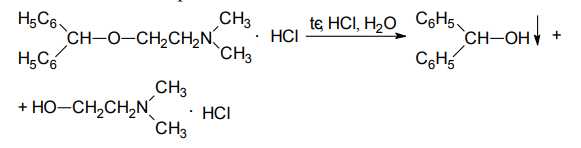 11 вопрос. Идентификация ЛВ по функц группам: 1) ионов цинка (р-ия с гексацианоферратом(П) →бел осадок) 2)ионов висмута (р-ия с сульфид-ионами→ черно-корич осадок) 2[ВiС l6]3- + 3S2- → Вi2S3 + 12Cl- 3)ионов бромида (действие нитрата серебра → желтовато-белый осадок) NaBr + AgNO3 = AgBr↓ + NaNO3 Br- + Ag+ = AgBr↓ 4)ионов карбоната -Р-ия с сульфатом магния Mg2+ + CO32- → MgCO3↓(белый) -Р-р карбоната (1:10) при приб 1 капли ф/ф раствора 1 % окраш в крас цвет (отличие от гидрокарбоната) Na2CO3 + HCL = NaCl + CO2 + H20 Выделяется СO2, кот при пропускании через Сa образует бел ос Сa(OH)2 + CO2 =CaCO3 + H2O 12 вопрос. Идентификация ЛВ по функц группам: 1) ионов магния MgSO4 + Na2HPO4 + NH4OH → MgNH4PO4↓ + Na2SO4 + H2O (бел крист осадок) 2) ионов аммония (р-ия разложения) NH4Cl + NaOH →NH3↑ + NaCl + H2O 3)ионов хлорида (действие р-ра нитрата серебра → бел творожистый ↓) NaCl + AgNO3 = AgCl↓ + NaNO3 Cl- + Ag+ = AgCl↓ 4) ионов нитрата -неск кристаллов антипирина растворяют в 2 к HCl, приб р-ор нитрита →зел окр-ие (отличие от н 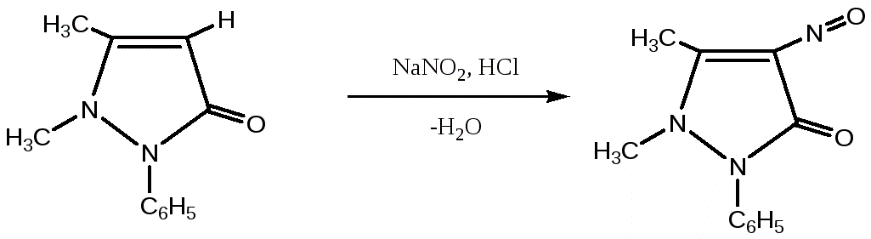 итратов). итратов).13 вопрос. Идентификация ЛВ по функц группам: 1) ионов серебра AgNO3+HCl→ AgCl↓(белый) +HNO3 2)ионов натрия NaCl + K[Sb(OH)6] → Na[Sb(OH)6] ↓(белый) + KCl Соль натрия, смоч HCl 25 % и внесенная в бесцв пламя, окр его в желтый цвет. 3)ионов иодида -NaI + AgNO3 (прис. HNO3) → AgI ↓+ NaNO3 желт творож -Проба Бельштейна. В-во на медной проволоке вносят в пламя, окр его в зел цвет. 4)ионов фосфата - Na3PO4 + 3AgNO3 → Ag3PO4↓(желтый) +3 NaNO3 - Na3PO4 + 3NH4CI + NH3. Н2О + МgSO4 → NH4МgPO4¯↓(белый) + 3NaCl + (NH4)2SO4, раствор в развед мин к-тах 14 вопрос. Идентификация ЛВ по функц группам: 1) ионов калия KCI + H2C4H4O6 → KHC4H4O6↓(белый) + HCI 2)ионов кальция CaCl2 + (NH₄)₂C₂O₄→СаС2О4(белый)+NH₄NO3 4)ионовнитрата2NaNO3 + H2SO4 → Na2SO4+ 2HNO3 Cu + HNO3 → Cu(NO3)2 + NO2↑(бурый) + H2O 3)ионов бромида 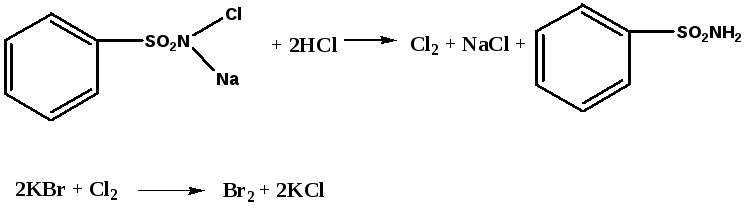 (хлороформ слой окр в желто-бурый (хлороформ слой окр в желто-бурый15 вопрос. Идентификация ЛВ по функц группам: 1)ионов железа Железо(II) FеСl2 + K3[Fe(CN)6] → 2КСl + KFe(CN)6¯(синий) Железо(III) Fe3+ + 6CNS- → [Fe(CNS)6]3- (красный) 2)ионов магния МgCl2 + Na2HPO4 + NH3 → (NH4Cl) → NH4МgPO4¯(белый)+ 2 NaCl 3) ионов хлорида HCl + AgNO3→ AgCl↓(белый) + HNO3 4) ионов фторида (с р-ром Fe(III) тиационата) -Fe(SCN)3 + 6NaF → Na3[FeF6](бесцветный) + 3NaSCN -2NaF + CaCl2 → CaF2 ↓(белый) + 2NaCl 16 вопрос. Идентификация орг ЛВ физико-хим методами: ИК с-ия. Спектры поглощения в вид и УФ-областях, возник в результате электр переходов в атомах и молекулах. Поглощение в ИК-области обусл переходами между колебат уровнями. Поглощение в УФ- и видим областях. Спектры поглощения в УФ- и видимой областях содержат как качеств, так и количеств информацию о поглощ в-ве. З-н Бугера-Ламберта-Бера A=k*C*l, А – опт плотность раствора; l – толщина поглощ слоя, см; С – конц-ия р-ра. Образцы, исп в абсорбц спектроскопии в УФ- и видимой областях, – это разбавленные растворы. Хроматография – физ-хим метод разделения и анализа смесей газов, паров, жидкостей или раствор веществ сорбционными методами. Она осн на различ разделении компонентов смеси между 2 фазами – неподвиж и подвиж. Вещества, сост НФ -сорбент. ПФ (элюент) – это поток жидкости или газа, фильтрующ через слой сорбента. Тонкослойная х-ия — основ на исп тонкого слоя адсорбента в качестве НФ. Он основан на том, что разделяемые вещества по-разному распред между сорбирующим слоем и протекающим через него элюентом. ВЭЖХ. Основой явл участие компонентов разделяемой смеси в сложной системе Ван-дер-Ваальсовых взаимодействий на границе раздела фаз. ВЭЖХ входит в состав группы методов, которая, ввиду сложности исслед объектов, включает предвар разделение исходной сложной смеси на относительно простые. Получ простые смеси анализируются затем обычными физ-хим методами или спец методами, созданными для хроматографии. Принцип ЖХ состоит в разделении компонентов смеси, осн на различии в равновесном распределении их между 2 несмеш фазами, одна из которых неподвижна, а другая подвижна (элюент). ГХ — физ-хим метод разделения веществ, основ на распред компонентов анализир смеси между двумя несмеш и движущимися относительно друг друга фазами, где в качестве подвиж фазы выступает газ, неподвиж фазы - тв сорбент или жидкость, нанесен на инертный твердый носитель или внутр стенки колонки. От типа исп неподвижной фазы ГХ подразделяют на газоадсорбционную и газожидкостную хр-ию. В первом случае НФ является тв носитель (силикагель, уголь, оксид алюминия), во 2 — жидкость, нанесён на поверхность инертного носителя. 17. Показатель «Описание» Агрегат состояния (тв в-во, жидкость, газ), если твердое вещество, то опред степень его дисперсности (мелкокрист, крупнокрист), форма кристаллов (игольчатые, цилиндрические). Цвет (небольш кол-во вещества помещают тонким слоем на чашку Петри или час стекло и рассматривают на белом фоне). Метод. Исп проводят в одинак пробирках из бесцв, прозрач, нейтр стекла с внутр диаметром около 12 мм, используя равные объемы – 2,0 мл испытуемой жидкости и воды, или растворителя, или эталона сравнения. Сравнивают окраску при рассеянном дневном свете, горизонтально (перпендикулярно оси пробирок) на матово-белом фоне. Запах опред редко сразу после вскрытия упаковки на расстоянии 4-6 см. Отсутствие запаха после вскрытия упаковки сразу по методике: 1-2 г вещества равномерно распред на час стекле диаметром 6-8 см и через 2 мин опред запах на расстоянии 4-6 см. 18. Показатель «Плотность» Плотность - масса единицы объема вещества (отношение массы в-ва к его объему при 20 °С. Относит плотность явл отношением массы опред объема вещества к массе равного объема воды при 20 °С. Определение плотности проводят с помощью пикнометра, ареометра или плотномера. Метод 1. Чистый сухой пикнометр взвеш с точ до 0,0002 г, заполняют водой немного выше метки, закр пробкой и выдерж 20 мин в термостате при (20 ± 0,1) ºС. Уровень воды в пикнометре доводят до метки, отбирая излишек воды пипеткой. Снова закр пробкой и выдерж в термостате еще 10 мин. Затем пикнометр вынимают из термостата, проверяют положение мениска воды, кот должен находиться на ур метки. Вытирают фильтр бумагой внутр поверхность горлышка и весь пикнометр снаружи, закр пробкой. Выдерж пикнометр под стеклом анал весов 10 мин и взвеш. Пикнометр освобождают от воды, высуш, ополаскивая последоват спиртом и эфиром, удаляют остатки эфира продуванием воздуха, заполняют пикнометр испыт жидкостью и проводят те же операции, что и с водой. Метод 2. Для определения плотности тв жиров и воска пикнометром. Проводят все операции с водой и высуш пикнометр. Пипеткой вносят в пикнометр расплавл жир или воск, 1/3 – 1/2 от объема пикнометра. Пикнометр без пробки ставят на 1 ч в горяч воду, затем охлажд до 20 °С и взвеш. Содерж пикнометра доводят до метки водой при температуре 20 °С, вытирают пикнометр и снова взвешивают. В обеих фазах и на поверхности их раздела не должно быть пузырьков воздуха. Метод 3. Применяют для опред плотности жидкостей с точ до ± 0,01 г/см3 с пом ареометра. Испыт жидкость помещ в цилиндр и при 20 ºС осторожно опускают в нее сухой ареометр, на шкале кот предусмотрена ожид велич плотности. Ареометр не должен касаться стенок и дна цилиндра. Через 3 – 4 мин после погружения ареометра произв отсчет по делению шкалы ареометра, соотв ниж мениску жидкости. Метод 4 Применяют для определения плотности жидкостей и газов в малом объеме (1 – 2 мл) с точностью до ± 0,0001 плотномером. Основан на опред периода колебаний U-образной измерит трубки опред объема, вызываемых ЭМ генератором. Измер спец датчиком период колебаний измерит трубки автоматически пересчитывается на плотность образца в г/см3 . 19. Определение влаги и летучих в-в . 1. Метод К. Фишера (полумикрометод) Основан на хим взаимодействии воды с компонентами реактива Фишера (р-р серы диоксида, йода и пиридина в метаноле). Взаимодействие реактива с водой протекает в 2 стадии стехиометрически по уравнениям: H2O + SO2 + I2 + 3C5H5N →2C5H5N · HI + C5H5NSO3 C5H5NSO3 + CH3OH → C5H5N ⋅ HSO4CH3 Исп растворы и реактивы должны быть безводными .С пом реактива Фишера может быть определена как гигроскопическая, так и кристаллизац вода. При этом воду можно определять в орг и неорг соединениях, в различных растворителях и летучих веществах. Методика А. Точную навеску испыт вещества, содерж от 30 до 50 мг воды, помещ в сосуд для титрования, в кот предва внесено 5,0 мл метанола безводного. Перемеш 1 мин и титруют р-ом Фишера, прибавляя его при приближ к конечной точке по 0,1–0,05 мл. Параллельно проводят к.о. Методика Б. Около 20 мл метанола безводного, помещ в сосуд и титруют р-ом Фишера, определяя к.т.т. Затем в сосуд для титрования вносят точную навеску испыт вещества. Смесь перемеш 1 мин и снова титруют р-ом Фишера, определяя к.т.т. амперометрически. Методика В. Около 10 мл метанола безводного или растворителя, помещают в сосуд для титрования и титруют йодсернистым реактивом, определяя к.т.т. амперометрически. Затем быстро вносят в сосуд для титрования указ количество испыт вещества и точно отмеренный объем йодсернистого реактива, взятый с изб приблизительно на 1 мл или объем. Сосуд закр пробкой, выдерживают в защ от света месте в течение 1 мин или в течение времени, периодически перемешивая содержимое сосуда. Избыток йодсернистого реактива титруют до первонач значения силы тока, используя метанол безводный или растворитель, к кот было прибавлено точно известное количество воды, эквивалентное около 2,5 мг/мл. 2. Микрометод определения воды (кулонометрический) При кулонометрическом титровании необходимый для реакции Фишера йод образ при анодном окислении йодид-иона: 2J − − 2e → J2 Образующийся йод реагирует с присутств водой и диоксидом серы. Изб йода указывает на достижение к.т.т. Кол-во оттитрованной воды пропорционально количеству электричества, пропущенному через ячейку. 1 моль йода соотв 1 молю воды, а кол-во электричества 10,71 Кл соотв 1 мг воды. Вследствие малого тока титр-ия кулонометрическое опр-ие применяется для количеств определения микроколичеств воды. Главным блоком прибора является кулонометрическая ячейка. Каждое отделение содержит платиновый электрод. Отделения разделены диафрагмой, предотвр смешение 2 растворов. Влагу удаляют из системы предвар электролизом. Катодное отделение также должно быть безводным. Анализир жидкая проба вводится в ячейку с анолитом шприцем через силиконовую прокладку. Следует избегать ввода твердых проб в ячейку. Газы вводятся в анолит через трубку для ввода газа. Объем пробы не должен превышать 10 мл. 3. Определение воды методом дистилляции Определение в приборе, сост из стекл круглодонной колбы вм от 250 до 500 мл, приемника, представл собой градуир пробирку или бюретку вм 6–10 мл, и холодильника. Методика В колбу отвеш испыт в-ва, приб 100 мл толуола или ксилола и неск кусочков пористого материала. Кипячение прекращают, когда объем воды в приемнике перестанет увеличиваться и верхний слой растворителя в приемнике станет прозрачным. Внутр трубку холодильника промывают толуолом и нагревают еще 5 мин, после чего приемник охлаждают до комнат t и стряхивают со стенок приемника все капли воды. Вся отогнанная вода собир в ниж части приемника. После полного разделения слоев отмечают объем отогнанной воды. 20. Показатели «Прозрачность» и «Цветность» Определение окраски жидкости опред визуально путем сравнения с соотв эталонами. Испытуемые жидкости и эталоны берут для сравнения в равных количествах. Сравнение проводят по общим правилам сравнения окраски: пробирки должны быть одинак стекла и диаметра; наблюд проводят при дневном отраженном свете на матово-белом фоне визуально путем сравнения с соответствующими эталонами; бесцв считаются жидкости, кот по цвету не отличаются от воды, а в случае растворов – от соотв растворителя. Бесцв р-р рассматр сверху через весь слой жидкости на матово-белом фоне. Для эталонов цветности вначале готовят 1) исходные растворы: р-р А (хлорида кобальта) CoCl2*6H2O – розовый; р-р Б (дихромата калия) K2Cr2O7 - желто-оранжевый; р-р В (сульфата меди) CuSO4*5H2O – синий; р-р Г (хлорида железа) FeCl3*6H2O – желто-бурый Эталоны цветности для сравнения приготавливают из осн растворов путем разбавления их раствором серной кислоты (0,1 моль/л). Получ по 7 эталонов в каждой из четырех шкал оттенков (а, б, в, г).: коричневых, желтых, розовых, зеленых. В качестве растворителя для приготовления основных растворов и эталонов используют 0,1 моль/л раствор серной кислоты. Определение прозрачности и степени мутности растворов. Прозрачность и степень мутности опред путем сравнения исслед р-ра с растворителем (если жидкость должна быть прозрачной) или эталоном (если опред степень мутности). Жидкость считают прозрачной, если при ее рассмотрении невооруж глазом не наблюд присутствие нераствор частиц. Сравнение проводят по общим правилам сравнения прозрачности и степени мутности: для сравнения берут равные объёмы испыту жидкости и эталонного раствора (5 или 10 мл); пробирки должны быть с притёртыми пробками один стекла и диаметра; наблюдение проводят при освещении электр лампой матового стекла мощностью 40 Вт на черном фоне при вертик расположении пробирок (смотрят сбоку). талонами по ГФ служат взвеси в воде из исходных веществ - гидразина сульфата и гексаметилентетрамина (метенамина). Эталон р-ры (I, II, III, IV) готовят из осн эталона разведением водой очищенной. 21. «Зола» Величина зольного остатка позволяет судить о загрязненности примесями, дающими при сжигании минеральный (зольный) остаток. Виды золы - общая зола, сульфатная зола. Определение сульфатной золы. Фарфоровый, кварцевый или платиновый тигель прокаливают при 550 – 650 ºС в теч 30 мин, охлаждают в эксикаторе над силикагелем или др подходящим осушителем и точно взвешивают по окончании каждого прокаливания. Точную навеску (обычно 1 – 2 г) помещ в предвар прокаленный тигель, смач 1 мл серной кислоты конц и осторожно (избегая сильного вспенивания вещества) нагревают на пламени, с закрт нагреват элементом и терморегулятором до обугливания. После охлаждения смач остаток 1 мл серной кислоты конц и нагревают до удаления паров серной кислоты. Затем тигель помещ в муфел печь и прокаливают при 550 – 650 ºС до тех пор, пока остаток полностью не превратится в пепел. По окончании прокаливания тигель охлажд в эксикаторе, взвешивают и рассчит процентное содержание остатка. В случае получения результата, превышающего допустимый предел - остаток вновь смачивают серной кислотой конц, сжигают в течение 30 мин, прокаливают до пост массы. 22. Значение критерия «Растворимость» Растворимость-приблиз растворимость фарм субст и вспом в-в при фиксир температуре. Испытание при (20 ± 2) ºC. Явл-я показателем чистоты вещества. Методика: к навеске растрерт.в-ва+р-ль-->встряхивают(10 мин при t=20+-2°). Для медленно р-ых в-в допуск нагрев на вод.бане до 30° далее встряхиваем 1-2 мин. Усл.обозначения: оч.легко р-им-до 1 мл; легко р-им- от 1 до 10; р-им- от 10 до 30; умеренно- от 10 до 100; мало-от 100 до 1000; оч.мало-от 1000 до 10000; практич.нераств- более 10000. 23. Источники и причины загрязнения ЛВ. Качество ЛП опред установлением его подлинности, определением его чистоты и количеств содержанием чистого вещества в препарате. Источники: провизор, плохая очистка сырья, побоч продукты синтеза, мех загряз, остатки р-лей (спирт,вода), аппаратура, наруш усл хранения. ГФ либо требует полного отсутствия примесей, либо допуск опред для данного препарата максимально допустимый предел примесей, кот не влияет на качество препарата и его лечебный эффект. Результат реакции на ту или другую примесь в испытуемом препарате сравнивается с результатом реакции, проведенной с теми же реактивами и в том же объеме с эталонным, стандартным, раствором, содержащим допустимое количество примеси. Сопоставление исследуемых растворов со стандартным дает возможность судить об отсутствии или наличии примеси в большем или меньшем количестве по сравнению с эталоном. В фармакопейном анализе часто при описании реакции указывается время, в течение которого необходимо вести наблюдение за происходящей реакцией. Только при условии соблюдения всех требований фармакопеи к анализу препаратов можно быть уверенным в его доброкачественности. Категории: зола общ и не р-мая в HCl, влажность, микроб чистота, частиц проходящих сквозь сито с опред.размером, минер и органич.примесь.,соли тяж металлов. Специфические, то есть имеющие собственное токсическое действие, или искажающие действие основного вещества: ионы-антагонисты по фармаколог действию, исходные вещества и промежуточные продукты синтеза препаратов, параллельно экстрагируемые вещества -сопутствующие вещества, имеющие близкие свойства и структуру, но отличные по фармаколог действию, вещества, образующиеся в результате длит или неправильного хранения. Общие примеси: хлориды, сульфаты, соединения кальция, цинка, железа, тяжёлых металлов, мышьяка, соли аммония. Все примеси могут быть допустимыми и недопустимыми. Испытание на недопустимые проводится обязательно, результат должен быть однозначно отриц. ГФ использует 2 способа определения содержания примесей: эталонный (если установлена ФС ПДК, например, в препарате «Меди сульфат» хлоридов должно быть не более 0,005%) и безэталонный (если примеси не должно быть). 24. Комплексонометрия в анализе солей неорг и орг ЛВ В основе комплексонометрии лежат реакции образования прочных и хорошо растворимых в воде внутрикомплексных соединений ионов металлов с комплексонами. Комплексоны – органические соединения – аминополикарбоновые кислоты и их соли. ТИТРАНТ – ЭТИЛЕНДИАМИНТЕТРААЦЕТАТА ДИНАТРИЕВАЯ СОЛЬ (ТРИЛОН Б, комплексон III); ИНДИКАТОР – ЭРИОХРОМ ЧЁРНЫЙ (металлохромный); СРЕДА – аммиачный буферный раствор нейтрализует выделяющуюся неорганическую кислоту, предупреждая обратимость реакций; П 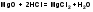 РЯМОЕ ТИТРОВАНИЕ. Магния оксид количественно определяют после растворения в минеральных кислотах: РЯМОЕ ТИТРОВАНИЕ. Магния оксид количественно определяют после растворения в минеральных кислотах:  Титр с синего → красно-фиол. 25. Методы аргентометрии А) Метод Мора: прямое титрование Определяем :Cl- ;Br– (I- не титурют этим методом, тк КТТ наступает раньше). Титрант:AgNO3, индикатор:K2CrO4 NaCI + AgNO3 → AgCI↓ + NaNO3 NaBr + AgNO3 → AgBr↓+ NaNO3 Реакция индикации: 2AgNO3 + K2CrO4 → Ag2CrO4↓(оранж-красн)+ 2KNO3 Б)Метод Фаянса: титрант-AgNO3, индикаторы – адсорбционные(натрия эозиат –определяет Br – ;I), среда :уксусо-кислая. Индикатор: бромфеноловый синий (в КТТ сине-фиолетовый) KI+ AgNO3→AgI↓+KNO3 до т.э.(AgI)I- после т.э. (AgI)Ag+ →((Ag)Ag+)*Ind-, титруют до изменения цвета осадка. В) Фольгарда (обратное титрование). Галогены осаждаются избытком титранта серебра нитрата. Не вступивший в реакцию осаждения серебра нитрат, оттитровывается р-ром тиоцианата аммония, индикатор - железоаммонийные квасцы. NaCI +AgNO3(избыток) → AgCI↓ + NaNO3 AgNO3(остаок) +NH4SCN →AgSCN↓ +NH4NO3 Реакция индикации: NH4SCN +NH4Fe(SO4)2→ Fe(SCN)3 +(NH4)2 SO4 (красное окрашив) В. Косвенный метод Фольгарда(прямое титрование)-к опреде веществу добавл 0,1 мг тиоционата аммония, титрат: AgNO3, инд:железоаммонийные квасцы Fe(SCN)3 NH4SCN +NH4Fe(SO4)2→ Fe(SCN)3 +(NH4)2 SO4 KI+ AgNO3→AgI↓+KNO3 3 AgNO3 + Fe(SCN)3→ 3AgSCN↓+Fe(NO3)3 (раствор обесцвечивается) 26. Методы Къельдаля Къельдаля (для определение азота в орг соединениях). Определение проводят в приборе для определения азота по методу Къельдаля. Стеклянная посуда должна быть термостойкой. Метод основан на минерализации органических ЛВ серной кислотой концентрированной (иногда в присутствии катализаторов) при нагревании. Минерализацию проводят в течение нескольких часов до получения раствора светло-зеленого цвета. В результате азот превращается в аммония сульфат. Затем добавляют раствор натрия гидроксида. Образующийся аммиак перегоняют с паром в колбу-приемник, содержащий для его поглощения, в зависимости от варианта титрования, соответствующую поглощающую жидкость: – для варианта прямого титрования – раствор борной кислоты; – для варианта обратного титрования – титрованные растворы серной или хлористоводородной кислоты. Перегонку ведут до получения 100 мл отгона в колбе-приемнике. В методе прямого титрования поглощенный аммиак титруют титрованным раствором хлористоводородной кислоты; в методе обратного титрования – титрованным раствором гидроксида натрия. По результатам титрования рассчитывают количественное содержание АЗОТА (g, %) в определяемой субстанции по формулам: – для прямого титрования: g= k*T*V*100 / а*1000 где V – объем титрованного раствора, пошедший на титрование, мл; K – поправочный коэффициент титрованного раствора; ТВ/А – титр рабочего раствора (титранта) по определяемому веществу, г/мл; а – навеска анализируемого вещества, взятая на анализ, г; – для обратного титрования: g=(V1*K1-V2*K2)*T*100 / а*1000 где V1; V2 – объемы титрованных растворов, соответственно взятого в избытке и затраченного на титрование, мл; К1; К2 – соответственно поправочные коэффициенты использованных титрованных растворов. 1. метод Къельдаля (классический) 1. В колбе Къельдаля: 1) 2RN + H2SO4 → (NH4)2SO4 + . . . 2) (NH4)2SO4 + 2NaOH → 2NH4OH + 3) Na2SO4 2NH4OH → 2NH3↑ + 2H2O 2  . В колбе приемнике: . В колбе приемнике:  1.3.Титрование Индикатор – смешанный (метиловый оранжевый и метиленовая синь): титруют до перехода окраски от зеленой до красно-фиолетовой). 2. Модифицированный метод Къельдаля: основан на разложении пирацетама с выделением аммиака при нагревании с раствором гидроксида натрия. 2  .1. Колба Къельдаля: .1. Колба Къельдаля: 2  .2. Колба – приёмник .2. Колба – приёмник 2  .3.Титрование .3.Титрование27. Кислотно-основное титрование слабых кислот в неводных растворителях на примере сульфаниламидов В СРЕДЕ ДМФА – согласно НД определяют фталазол, салазопиридазин. Индикатор – тимоловый синий (бесцветный – синий). Параллельно – контрольный опыт на индикатор: f   экв (А) = ½ экв (А) = ½28. Кислотно-основное титрование слабых кислот в неводных растворителях на примере барбитуровой кислоты А  цидиметрия (в среде ДМФА): титрант – раствор едкого натра в смеси метанола и бензола или раствор метилата натрия, индикатор – тимоловый синий: цидиметрия (в среде ДМФА): титрант – раствор едкого натра в смеси метанола и бензола или раствор метилата натрия, индикатор – тимоловый синий:  29. Кислотно-основное титрование в водных и смешанных растворителях на примере салициловой кислоты, натрия салицилата. В  ВОДНОЙ СРЕДЕ: Растворитель – вода; титруют в присутствии эфира (для извлечения образующихся кислот, которые изменяют рН среды и изменяют окраску индикатора раньше наступления точки конца титрования); титрант – раствор хлороводородной кислоты; индикатор – смешанный (смесь равных количеств метилового оранжевого и метиленового синего): ВОДНОЙ СРЕДЕ: Растворитель – вода; титруют в присутствии эфира (для извлечения образующихся кислот, которые изменяют рН среды и изменяют окраску индикатора раньше наступления точки конца титрования); титрант – раствор хлороводородной кислоты; индикатор – смешанный (смесь равных количеств метилового оранжевого и метиленового синего):В НЕВОДНОЙ СРЕДЕ: Растворитель - ледяная уксусная кислота; титрант – хлорная кислота; индикатор – кристаллический фиолетовый:  30. Йодометрия и йодхлорметрия на примере фенолов 1. Йодометрия в присутствии натрия гидрокарбоната или натрия ацетата для связывания выделяющегося йодоводорода.  NaHCO3 + HI → NaI + CO2 + H2O, до синего окр-ия 2  . Метод обратной иодхлорметрии (иодиметрии): . Метод обратной иодхлорметрии (иодиметрии): 31. Броматометрия на примере фенолов 1. Метод прямой броматометрии (тимол) в сернокислой среде, индикатор – м/о:  2. Метод обратной иодиметрии (резорцин, фенол) с иодиметрическим окончанием:   32. Нитритометрия в анализе ЛВ. Нитритометрия – метод титриметр анализа, при котором в качестве титрованного раствора используется раствор натрия нитрита. Применяется для количеств определения соединений, содержащих первич или вторич ароматическую аминогруппу, для определения гидразидов, а также арома нитросоединений после предвар восстановления нитрогруппы до аминогруппы. Методика. Точную навеску растворяют в смеси 10 мл воды и 10 мл хлористоводородной кислоты развед 8,3 %. Прибавляют воду до общего объема 80 мл, 1 г калия бромида и при пост перемешивании титруют натрия нитрита раствором 0,1 М. В начале титрования нитрита со скоростью 2 мл/мин, а в конце – 0,05 мл/мин. Титрование проводят при t раствора 15 — 20 ºС, однако в некоторых случаях требуется охлаждение до 0 — 5 ºС. 33. Методики определения д 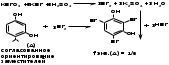 опустимых и недопустимых примеси хлоридов эталонным и безэталонным способами. опустимых и недопустимых примеси хлоридов эталонным и безэталонным способами. Растворы хлоридов в зависимости от их концентрации образуют с раствором нитрата серебра белый творожистый осадок, белую муть или опалесценцию, не исчезающие от прибавления азотной кислоты и легко исчезающие от прибавления раствора аммиака. NaCl + AgNO3 → NaNO3 AgCl. Методика: 10 мл раствора испыт препарата, приготовленного прибавляют 0,5 мл азотной кислоты, 0,5 мл раствора нитрата серебра, перемешивают и через 5 мин сравнивают с эталоном, состоящим из 10 мл эталонного раствора Б и такого же количества реактивов, какое прибавлено к испытуемому раствору. Опалесценция, появившаяся в испытуемом растворе, не должна превышать эталон. 34. Методики определения допустимых и недопустимых примеси сульфатов эталонным и безэталонным способами. Растворы сульфатов в зависимости от их концентрации образуют с растворами солей бария белый осадок или муть, не исчезающие от прибавления разведенной хлористоводородной кислоты. SO42- + Ba2+ BaSO4↓ белыйосадок Методика: к 10 мл раствора испыт препарата, приготовленного прибавляют 0,5 мл разведенной хлористоводородной кислоты и 1 мл раствора хлорида бария, перемешивают и через 10 мин сравнивают с эталоном, состоящим из 10 мл эталонного раствора Б и такого же количества реактивов, какое прибавлено к испытуемому раствору. Муть, появившаяся в испытуемом растворе, не должна превышать эталон. 35. Методики определения допустимых и недопустимых примеси солей аммония эталонным и безэталонным способами. МЕТОД 1. Растворы солей аммония в зависимости от их концентрации образуют с реактивом Несслера желто-бурый осадок или желтое окрашивание. NН4Сl+ 2 К2[HgI4] + 4 КОН = [ОHg2NН2 +7КI +КСl +3 Н] I2О Методика: к 10 мл раствора испытуемого препарата, приготовленного, как указано в соответствующей частной статье, прибавляют 0,15 мл реактива Несслера, перемешивают и через 5 мин сравнивают с эталоном состоящим из 10 мл эталонного раствора Б и такого же количества реактива, какое прибавлено к испытуемому раствору. Окраска, появившаяся в испытуемом растворе, не должна превышать эталон. МЕТОД 2. Соли аммония при прибавлении едкого натра выделяют аммиак, который определяют по запаху или посинению смоченной водой красной лакмусовой бумаги. NН4Сl+ NаОН = NН3↑ + Н2О + NаСl Методика: 5 мл раствора испытуемого препарата, указанной в соответствующей частной статье концентрации, помещают в коническую колбу вместимостью 25 мл, прибавляют 5 мл раствора едкого натра. Сверху колбы помещают смоченную водой красную лакмусовую бумагу и закрывают часовым стеклом. Колбу ставят на водяную баню. Наблюдение проводят через 5 мин. 36. Методики определения допустимых и недопустимых примеси солей кальция эталонным и безэталонным способами. Растворы солей кальция в зависимости от их концентрации дают с раствором оксалата аммония белый мелкокристаллический осадок или белую муть, не исчезающую от прибавления уксусной кислоты, но легко растворимые при прибавлении хлористоводородной или азотной кислоты. CaСl2 + (NH4) 2C2O4 → СаС2O4 + 2NH4Cl Определение кальция в неорганических соединениях. К 10 мл раствора испытуемого препарата, приготовленного прибавляют 1 мл раствора хлорида аммония, 1 мл раствора аммиака и 1 мл раствора оксалата аммония, перемешивают и через 10 мин сравнивают с эталоном, состоящим из 10 мл эталонного раствора Б и такого же количества реактивов, какое прибавлено к испытуемому раствору. Муть, появившаяся в испытуемом растворе, не должна превышать эталон. 37. Методики определения допустимых и недопустимых примеси солей железа эталонным и безэталонным способами. Растворы солей двух- и трехвалентного железа в зависимости от концентрации образуют с раствором сульфосалициловой кислоты в аммиачной среде коричнево-красные или желтые растворы феррилсульфосалицилатных комплексов.  Методика: к 10 мл раствора испытуемого препарата, приготовленного, как указано в соответствующей частной статье, прибавляют 2 мл раствора сульфосалициловой кислоты и 1 мл раствора аммиака и через 5 мин сравнивают с эталоном, состоящим из 10 мл эталонного раствора В и такого же количества реактивов, какое прибавлено к испытуемому раствору. Окраска, появившаяся в испытуемом растворе, не должна превышать эталон. 38. Методики определения допустимых и недопустимых примеси солей цинка эталонным и безэталонным способами. Растворы солей цинка в зависимости от концентрации образуют с раствором ферроцианида калия белый осадок или муть, нерастворимые в разведенных кислотах. Zn(NO3)2 + K4[Fe(CN)6] Методика: к 10 мл раствора испытуемого препарата, приготовленного, как указано в соответствующей частной статье, прибавляют 2 мл хлористоводородной кислоты, 5 капель раствора ферроцианида калия и через 10 мин сравнивают с эталоном, состоящим из 10 мл эталонного раствора Б и такого же количества реактивов, какое прибавлено к испытуемому раствору. Муть, появившаяся в испытуемом растворе, не должна превышать эталон. 39. Методики определения допустимых и недопустимых примеси солей тяжелых металлов эталонным и безэталонным способами. Растворы солей свинца в зависимости от концентрации образуют с растворами сульфида натрия или сероводорода черный осадок или бурое окрашивание раствора. Pb(NO3)2+ Na2S →PbS↓ + 2NaNO3 Методика: к 10 мл раствора испыт препарата прибавляют 1 мл разведенной уксусной кислоты, 2 капли раствора сульфида натрия, перемешивают и через 1 мин сравнивают с эталоном, состоящим из 1 мл эталонного раствора Б, такого же количества реактивов, какое прибавлено к испытуемому раствору, и 9 мл воды. Наблюдение окраски проводят по оси пробирок диаметром около 1,5 см, помещенных на белой поверхности. В сравниваемых растворах допустима лишь слабая опалесценция от серы, выделяющейся из сульфида натрия. |