Ответы по ПА. 3. Процессы адаптации. Виды адаптационных изменений
 Скачать 1.18 Mb. Скачать 1.18 Mb.
|

|
Причины жировой дистрофии разнообразны. Чаще всего она связана с кислородным голоданием (тканевая гипоксия), поэтому жировая дистрофия так часто встречается при заболеваниях сердечно-сосудистой системы, хронических заболеваниях легких, анемиях, хроническом алкоголизме и т.д. В условиях гипоксии страдают в первую очередь отделы органа, находящиеся в функциональном напряжении. Вторая причина - инфекции (дифтерия, туберкулез, сепсис) и интоксикации (фосфор, мышьяк, хлороформ), ведущие к нарушениям обмена (диспротеиноз, гипопротеинемия, гиперхолестеринемия), третья - авитаминозы и одностороннее (с недостаточным содержанием белков) питание, сопровождающееся дефицитом ферментов и липотропных факторов, которые необходимы для нормального жирового обмена клетки. Исход жировой дистрофии зависит от ее степени. Если она не сопровождается грубым поломом клеточных структур, то, как правило, оказывается обратимой. Глубокое нарушение обмена клеточных липидов в большинстве случаев заканчивается гибелью клетки, функция органов при этом резко нарушается, а в ряде случаев и выпадает. Системные липидозы (наследственные ферментопатии, болезни на- копления, лизосомные болезни)
8.Диспротеинозы: причины, патогенез, морфогенез, клинико-морфологическая характеристика, исходы Паренхиматозные белковые дистрофии (диспротеинозы) Большая часть белков цитоплазмы (простых и сложных) находится в соединении с липидами, образуя липопротеидные комплексы. Эти комплексы составляют основу мембран митохондрий, эндоплазматической сети, пластинчатого комплекса и других структур. Помимо связанных белков, в цитоплазме содержатся и свободные. Многие из последних обладают функцией ферментов. Сущность паренхиматозных диспротеинозов состоит в изменении физико-химических и морфологических свойств белков клетки: они подвергаются денатурации и коагуляции или, наоборот, колликвации, что ведет к гидратации цитоплазмы; в тех случаях, когда нарушаются связи белков с липидами, возникает деструкция мембранных структур клетки. В исходе этих нарушений может развиться коагуляционный (сухой) или колликвационный (влажный) некроз (схема I). К паренхиматозным диспротеинозам относят гиалиново-капельную, гидропическую и роговую дистрофии. К паренхиматозным белковым дистрофиям со времен Р. Вирхова причисляли и многие патологи продолжают причислять так называемую зернистую дистрофию, при которой в клетках паренхиматозных органов появляются белковые зерна. Сами органы увеличиваются в размерах, становятся дряблыми и тусклыми на разрезе, что послужило причиной называть также зернистую дистрофию тусклым (мутным) набуханием. Однако электронно-микроскопическое и гистоферменто-химическое изучение «зернистой дистрофии» показало, что в ее основе лежит не накопление белка в цитоплазме, а гиперплазия ультраструктур клеток паренхиматозных органов как выражение функционального напряжения этих органов в ответ на различные воздействия; гиперплазированные ультраструктуры клетки выявляются при светооптическом исследовании как белковые гранулы. 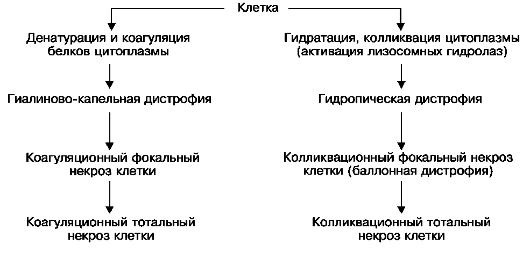 Схема I. Морфогенез паренхиматозных диспротеинозов Гиалиново-капельная дистрофия При гиалиново-капельной дистрофии в цитоплазме появляются крупные гиалиноподобные белковые капли, сливающиеся между собой и заполняющие тело клетки; при этом происходит деструкция ультраструктурных элементов клетки. В ряде случаев гиалиново-капельная дистрофия завершается фокальным коагуляционным некрозом клетки. Этот вид диспротеиноза часто встречается в почках, редко - в печени и совсем редко - в миокарде. В почках при микроскопическом исследовании накопление гиалиновых капель находят в нефроцитах. При этом наблюдается деструкция митохондрий, эндоплазматической сети, щеточной каемки . В основе гиалиново-капельной дистрофии нефроцитов лежит недостаточность вакуолярно-лизосомального аппарата эпителия проксимальных канальцев, в норме реабсорбирующего белки. Поэтому этот вид дистрофии нефроцитов очень часто встречается при нефротическом синдроме. Этот синдром является одним из проявлений многих заболеваний почек, при которых первично поражается гломерулярный фильтр (гломерулонефрит, амилоидоз почек, парапротеинемическая нефропатия и др.). Внешний вид почек при этой дистрофии не имеет каких-либо характерных черт, он определяется прежде всего особенностями основного заболевания (гломерулонефрит, амилоидоз). 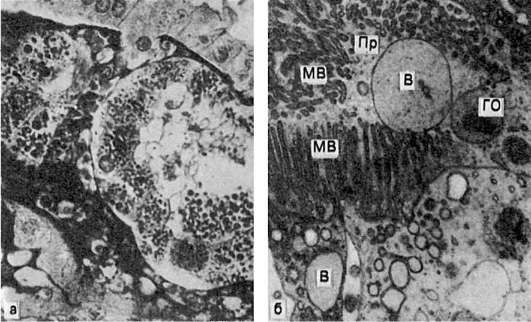 Гиалиново-капельная дистрофия эпителия почечных канальцев: а - в цитоплазме эпителия крупные белковые капли (микроскопическая картина); б - в цитоплазме клетки много белковых (гиалиновых) образований (ГО) овальной формы и вакуолей (В); отмечаются десквамация микроворсинок (МВ) щеточной каемки и выход в просвет (Пр) канальца вакуолей и белковых образований. Электронограмма. х18 000 Исход гиалиново-капельной дистрофии неблагоприятен: она завершается необратимым процессом, ведущим к некрозу клетки. Функциональное значение этой дистрофии очень велико. С гиалиново- капельной дистрофией эпителия почечных канальцев связаны появление в моче белка (протеинурия) и цилиндров (цилиндрурия), потеря белков плазмы (гипопротеинемия), нарушение ее электролитного баланса. Гиалиново-капельная дистрофия гепатоцитов нередко является морфологической основой нарушений многих функций печени. Гидропическая дистрофия Гидропическая, или водяночная, дистрофия характеризуется появлением в клетке вакуолей, наполненных цитоплазматической жидкостью. Она наблюдается чаще в эпителии кожи и почечных канальцев, в гепатоцитах, мышечных и нервных клетках, а также в клетках коры надпочечников. Микроскопическая картина: паренхиматозные клетки увеличены в объеме, цитоплазма их заполнена вакуолями, содержащими прозрачную жидкость. Ядро смещается на периферию, иногда вакуолизируется или сморщивается. Прогрессирование этих изменений приводит к распаду ультраструктур клетки и переполнению клетки водой. Клетка превращается в заполненные жидкостью баллоны или в огромную вакуоль, в которой плавает пузырьковидное ядро. Такие изменения клетки, которые по существу являются выражением фокального колликвационного некроза называют баллонной дистрофией. Внешний вид органов и тканей мало изменяется при гидропическои дистрофии, она обнаруживается обычно под микроскопом. Механизм развития гидропическои дистрофии сложен и отражает нарушения водно-электролитного и белкового обмена, ведущие к изменению коллоидно-осмотического давления в клетке. Большую роль играет нарушение проницаемости мембран клетки, сопровождающееся их распадом. Это ведет к закислению цитоплазмы, активации гидролитических ферментов лизосом, которые разрывают внутримолекулярные связи с присоединением воды. Причины развития гидропической дистрофии в разных органах неоднозначны. В почках - это повреждение гломерулярного фильтра (гломерулонефрит, амилоидоз, сахарный диабет), что ведет к гиперфильтрации и недостаточности ферментной системы базального лабиринта нефроцитов, в норме обеспечивающей реабсорбцию воды; поэтому гидропическая дистрофия нефроцитов так характерна для нефротического синдрома. В печени гидропическая дистрофия возникает при вирусном и токсическом гепатитах и нередко является причиной печеночной недостаточности. Причиной гидропическои дистрофии эпидермиса может быть инфекция (оспа), отек кожи различного механизма. Вакуолизация цитоплазмы может быть проявлением физиологической деятельности клетки, что отмечается, например, в ганглиозных клетках центральной и периферической нервной системы. Исход гидропической дистрофии, как правило, неблагоприятный; она завершается фокальным или тотальным некрозом клетки. Поэтому функция органов и тканей при гидропической дистрофии резко страдает 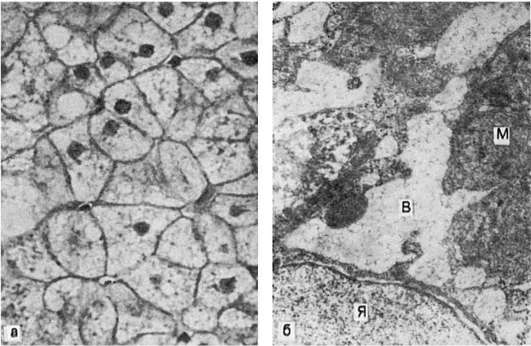 Гидропическая дистрофия печени (биопсия): а - микроскопическая картина; вакуолизация гепатоцитов; б - электронограмма: рас- ширение канальцев эндоплазматической сети и образование вакуолей (В), заполненных хлопьевидным содержимым. Мембраны, ограничивающие вакуоли, почти полностью ли- шены рибосом. Вакуоли сдавливают расположенные между ними митохондрии (М), часть которых подвергается деструкции; Я - ядро гепатоцита. х18 000 Роговая дистрофия Роговая дистрофия, или патологическое ороговение, характеризуется избыточным образованием рогового вещества в ороговевающем эпителии (гиперкератоз, ихтиоз) или образованием рогового вещества там, где в норме его не бывает (патологическое ороговение на слизистых оболочках, или лейкоплакия; образование «раковых жемчужин» в плоскоклеточном раке). Процесс может быть местным или распространенным. Причины роговой дистрофии разнообразны: нарушение развития кожи, хроническое воспаление, вирусные инфекции, авитаминозы и др. Исход может быть двояким: устранение вызывающей причины в начале процесса может привести к восстановлению ткани, однако в далеко зашедших случаях наступает гибель клеток. Значение роговой дистрофии определяется ее степенью, распространенностью и длительностью. Длительно существующее патологическое ороговение слизистой оболочки (лейкоплакия) может явиться источником развития раковой опухоли. Врожденный ихтиоз резкой степени, как правило, несовместим с жизнью. 9.Внутриклеточные накоплениягликогена: причины, патогенез, морфогенез, морфологическая характеристика и методы диагностики. Углеводные дистрофии, связанные с нарушением обмена гликогена Основные запасы гликогена находятся в печени и скелетных мышцах. Гликоген печени и мышц расходуется в зависимости от потребностей организма (лабильный гликоген). Гликоген нервных клеток, проводящей системы сердца, аорты, эндотелия, эпителиальных покровов, слизистой оболочки матки, соединительной ткани, эмбриональных тканей, хряща и лейкоцитов является необходимым компонентом клеток, и его содержа- ние не подвергается заметным колебаниям (стабильный гликоген). Однако деление гликогена на лабильный и стабильный условно. Регуляция обмена углеводов осуществляется нейроэндокринным путем. Основная роль принадлежит гипоталамической области, гипофизу (АКТГ, тиреотропный, соматотропный гормоны), (β-клеткам поджелудочной железы (инсулин), надпочечникам (глюкокортикоиды, адреналин) и щитовидной железе. Нарушения содержания гликогена проявляются в уменьшении или увеличении количества его в тканях и появлении там, где он обычно не выявляется. Эти нарушения наиболее ярко выражены при сахарном диабете и при наследственных углеводных дистрофиях - гликогенозах. При сахарном диабете, развитие которого связывают с патологией β-клеток островков поджелудочной железы, происходят недостаточное использование глюкозы тканями, увеличение ее содержания в крови (гипергликемия) и выведение с мочой (глюкозурия). Тканевые запасы гликогена резко уменьшаются. Это в первую очередь касается печени,в которой нарушается синтез гликогена, что ведет к инфильтрации ее жирами - развивается жировая дистрофия печени; при этом в ядрах гепатоцитов появляются включения гликогена, они становятся светлыми («дырчатые», «пустые», ядра). С глюкозурией связаны характерные изменения почек при диабете. Они выражаются в гликогенной инфильтрации эпителия канальцев, главным образом узкого и дистального сегментов. Эпителий становится высоким, со светлой пенистой цитоплазмой; зерна гликогена видны и в просвете канальцев. Эти изменения отражают состояние синтеза гликогена (полимеризация глюкозы) в канальцевом эпителии при резорбции богатого глюкозой ультрафильтрата плазмы. При диабете страдают не только почечные канальцы, но и клубочки, их капиллярные петли, базальная мембрана которых становится значительно более проницаемой для сахаров и белков плазмы. Возникает одно из проявлений диабетической микроангиопатии - интеркапиллярный (диабетический) гломерулосклероз. Наследственные углеводные дистрофии, в основе которых лежат нарушения обмена гликогена, называются гликогенозами. Гликогенозы обусловлены отсутствием или недостаточностью фермента, участвующего в расщеплении депонированного гликогена, и относятся поэтому к наследственным ферментопатиям, или болезням накопления. В настоящее время хорошо изучены 6 типов гликогенозов, обусловленных наследственной недостаточностью 6 различных ферментов. Это болезни Гирке (I тип), Помпе (II тип), Мак-Ардля (V тип) и Герса (VI тип), при которых структура накапливаемого в тканях гликогена не нарушена, и болезни Форбса-Кори (III тип) и Андерсена (IV тип), при которых она резко изменена Морфологическая диагностика гликогеноза того или иного типа возможна при биопсии с помощью гистоферментативных методов. 10.Нарушения обмена липофусцина и меланина: клинико-морфологическая характеристика. Нарушения обмена хромопротеидов (эндогенные пигментации)2 Хромопротеиды - окрашенные белки, или эндогенные пигменты, играют важную роль в жизни организма. С помощью хромопротеидов осуществляются дыхание (гемоглобин, цитохромы), выработка секретов (желчь) и инкретов (серотонин), защита организма от воздействия лучевой энергии (меланин), пополнение запасов железа (ферритин), баланс витаминов (липохромы) и т.д. Обмен пигментов регулируется вегетативной нервной системой, эндокринными железами, он тесно связан с функцией органов кроветворения и системы моноцитарных фагоцитов. Классификация. Эндогенные пигменты принято делить на 3 группы: -гемоглобиногенные, представляющие собой различные производные гемоглобина -протеиногенные, или тирозиногенные, связанные с обменом тирозина - липидогенные, или липопигменты, образующиеся при обмене жиров. Нарушения обмена протеиногенных (тирозиногенных) пигментов К протеиногенным (тирозиногенным) пигментам относят меланин, пигмент гранул энтерохромаффинных клеток и адренохром. Накопление этих пигментов в тканях служит проявлением ряда заболеваний. Меланин (от греч. melas - черный) - широко распространенный буро-черный пигмент, с которым у человека связана окраска кожи, волос, глаз. Он дает положительную аргентаффинную реакцию, т.е. обладает способностью восстанавливать аммиачный раствор нитрата серебра до металлического серебра. Эти реакции позволяют гистохимически отличить его в тканях от других пигментов. Синтез меланина происходит из тирозина в клетках меланинобразующей ткани - меланоцитах. Их предшественниками являются меланобласты. Под действием тирозиназы в меланосомах меланоцитов из тирозина образуется диоксифенилаланин (ДОФА), или промеланин, который полимеризуется в меланин. Клетки, фагоцитирующие меланин, называют меланофагами.  Кожа при аддисоновой болезни: а - в базальном слое эпидермиса скопления меланоцитов; в дерме много меланофагов; б - меланоцит кожи. В цитоплазме много меланосом. Я - ядро. Электронограмма. х10 000 Меланоциты и меланофаги содержатся в эпидермисе, дерме, радужной и сетчатой оболочках глаз, в мягкой мозговой оболочке. Содержание меланина в коже, сетчатке и радужке зависит от индивидуальных и расовых особенностей и подвергается колебаниям в различные периоды жизни. Регуляция меланогенеза осуществляется нервной системой и эндокринными железами. Стимулируют синтез меланина меланостимулирующий гормон гипофиза, АКТГ, половые гормоны, медиаторы симпатической нервной системы, тормозят - мелатонин и медиаторы парасимпатической нервной системы. Образование меланина стимулируется ультрафиолетовыми лучами, что объясняет возникновение загара как адаптивной защитной биологической реакции. Нарушения обмена меланина выражаются в усиленном его образовании или исчезновении. Эти нарушения имеют распространенный или местный характер и могут быть приобретенными или врожденными. Распространенный приобретенный гипермеланоз (меланодермия) особенно часто и резко выражен при аддисоновой болезни , обусловленной поражением надпочечников, чаще туберкулезной или опухолевой природы. Гиперпигментация кожи при этой болезни объясняется не столько тем, что при разрушении надпочечников вместо адреналина из тирозина и ДОФА синтезируется меланин, сколько усилением продукции АКТГ в ответ на уменьшение адреналина в крови. АКТГ стимулирует синтез меланина, в меланоцитах увеличивается количество меланосом. Меланодермия встречается также при эндокринных расстройствах (гипогонадизм, гипопитуитаризм), авитаминозах (пеллагра, цинга), кахексии, интоксикации углеводородами. Распространенный врожденный гипермеланоз (пигментная ксеродерма) связан с повышенной чувствительностью кожи к ультрафиолетовым лучам и выражается в пятнистой пигментации кожи с явлениями гиперкератоза и отека. К местному приобретенному меланозу относят меланоз толстой кишки, который встречается у людей, страдающих хроническим запором, гиперпигментированные участки кожи (черный акантоз) при аденомах гипофиза, гипертиреоидизме, сахарном диабете. Очаговое усиленное образование меланина наблюдается в пигментных пятнах (веснушки, лентиго) и в пигментных невусах. Из пигментных невусов могут возникать злокачественные опухоли - меланомы. Распространенный гипомеланоз, или альбинизм (от лат. albus - белый), связан с наследственной недостаточностью тирозиназы. Альбинизм проявляется отсутствием меланина в волосяных луковицах, эпидермисе и дерме, в сетчатке и радужке. Очаговый гипомеланоз (лейкодерма, или витилиго) возникает при нарушении нейроэндокринной регуляции меланогенеза (лепра, гиперпаратиреоидизм, сахарный диабет), образовании антител к меланину (зоб Хасимото), воспалительных и некротических поражениях кожи (сифилис). Нарушения обмена липидогенных пигментов (липопигментов) В эту группу входят жиробелковые пигменты - липофусцин, пигмент недостаточности витамина Е, цероид и липохромы. Липофусцин, пигмент недостаточности витамина Е и цероид имеют одинаковые физические и химические (гистохимические) свойства, что дает право считать их разновидностями одного пигмента - липофусцина. Однако в настоящее время липофусцином считают липопигмент лишь паренхиматозных и нервных клеток; пигмент недостаточности витамина Е - разновидность липофусцина. Цероидом называют липопигмент мезенхимальных клеток, главным образом макрофагов. Патология обмена липопигментов разнообразна. Липофусцин представляет собой гликолипопротеид. Он представлен зернами золотистого или коричневого цвета, электронно-микроскопически выявляется в виде электронно-плотных гранул , окруженных трехконтурной мембраной, которая содержит миелиноподобные структуры.  Липофусцин (Лф) в мышечной клетке сердца, тесно связанный с митохондриями (М). Мф - миофибриллы. Электронограмма. х21 000 Образование липофусцина происходит путем аутофагии и проходит несколько стадий. Первичные гранулы, или пропигмент-гранулы, появляются перинуклеарно в зоне наиболее активно протекающих обменных процессов. Они содержат ферменты митохондрий и рибосом (металлофлавопротеиды, цитохромы), связанные с липопротеидами их мембран. Пропигмент-гранулы поступают в пластинчатый комплекс, где происходит синтез гранул незрелого липофусцина, который суданофилен, ШИК-положителен, содержит железо, иногда медь, обладает светло-желтой аутофлюоресценцией в ультрафиолетовом свете. Гранулы незрелого пигмента перемещаются в периферическую зону клетки и абсорбируются там лизосомами; появляется зрелый липофусцин, обладающий высокой активностью лизосомных, а не дыхательных ферментов. Гранулы его становятся коричневыми, они стойко суданофильны, ШИК-положительны, железо в них не выявляется, аутофлюоресценция становится красно-коричневой. Накапливающийся в лизосомах липофусцин превращается в остаточные тельца - телолизосомы. В условиях патологии содержание липофусцина в клетках может резко увеличиваться. Это нарушение обмена называется липофусцинозом. Он может быть вторичным и первичным (наследственным). Вторичный липофусциноз развивается в старости, при истощающих заболеваниях, ведущих к кахексии (бурая атрофия миокарда, печени), при повышении функциональной нагрузки (липофусциноз миокарда при пороке сердца, печени - при язвенной болезни желудка и двенадцатиперстной кишки), при злоупотреблении некоторыми лекарствами (анальгетики), при недостаточности витамина Е (пигмент недостаточности витамина Е). Первичный (наследственный) липофусциноз характеризуется избирательным накоплением пигмента в клетках определенного органа или системы. Он проявляется в виде наследственного гепатоза, или доброкачественной гипербилирубинемии (синдромы Дабина-Джонсона, Жиль- бера, Кригера-Найяра) с избирательным липофусцинозом гепатоцитов, а также нейронального липофусциноза (синдром Бильшовского-Янского, Шпильмейера-Шегрена, Кафа), когда пигмент накапливается в нервных клетках, что сопровождается снижением интеллекта, судорогами, нарушением зрения. 11.Гемосидероз местный и системный. Гемохроматоз. Морфологическая характеристика и методы диагностики. Нарушения обмена гемоглобиногенных пигментов В норме гемоглобин проходит ряд циклических превращений, обеспечивающих его ресинтез и образование необходимых для организма продуктов. Эти превращения связаны со старением и разрушением эритроцитов (гемолиз, эритрофагия), постоянным обновлением эритроцитной массы. В результате физиологического распада эритроцитов и гемоглобина образуются пигменты ферритин, гемосидерин и билирубин. В патологических условиях вследствие многих причин гемолиз может быть резко усилен и осуществляться как в циркулирующей крови (интраваскулярно), так и в очагах кровоизлияний (экстраваскулярно). В этих условиях, помимо увеличения образующихся в норме гемоглобиногенных пигментов, может появляться ряд новых пигментов - гематоидин, гематины и порфирин. В связи с накоплением гемоглобиногенных пигментов в тканях могут возникать различные виды эндогенных пигментации, которые становятся проявлением ряда заболеваний и патологических состояний. Ферритин - железопротеид, содержащий до 23% железа. Железо ферритина связано с белком, который носит название апоферритина. В норме ферритин обладает дисульфидной группой. Это неактивная (окисленная) форма ферритина - SS-ферритин. При недостаточности кислорода происходит восстановление ферритина в активную форму - SH-ферритин, который обладает вазопаралитическими и гипотензивными свойствами. В зависимости от происхождения различают анаболический и катаболический ферритин. Анаболический ферритин образуется из железа, всасывающегося в кишечнике, катаболический - из железа гемолизированных эритроцитов. Ферритин (апоферритин) обладает антигенными свойствами. Ферритин образует берлинскую лазурь (железосинеродистое железо) под действием железосинеродистого калия и соляной, или хлористоводородной, кислоты (реакция Перлса) и может быть идентифицирован с помощью специфической антисыворотки при иммунофлюоресцентном исследовании. Большое количество ферритина содержится в печени (депо ферритина), селезенке, костном мозге и лимфатических узлах, где обмен его связан с синтезом гемосидерина, гемоглобина и цитохромов. В условиях патологии количество ферритина может увеличиваться как в тканях, так и в крови. Повышение содержания ферритина в тканях наблюдается при гемосидерозе, так как полимеризация ферритина ведет к образованию гемосидерина. Ферритинемией объясняют необратимость шока, сопровождающегося сосудистым коллапсом, так как SH-ферритин выступает в роли антагониста адреналина. Гемосидерин образуется при расщеплении гема и является полимером ферритина. Он представляет собой коллоидную гидроокись железа, связанную с белками, гликозаминогликанами и липидами клетки. Клетки, в которых образуется гемосидерин, называются сидеробластами. В их сидеросомах происходит синтез гранул гемосидерина . Сидеробласты могут быть как мезенхимальной, так и эпителиальной природы. Гемосидерин постоянно обнаруживается в ретикулярных и эндотелиальных клетках селезенки, печени, костного мозга, лимфатических узлах. В межклеточном веществе он подвергается фагоцитозу сидерофагами. Присутствие в гемосидерине железа позволяет выявлять его с помощью характерных реакций: образование берлинской лазури (реакция Перлса), турнбулевой сини (обработка срезов сульфидом аммония, а затем железо- синеродистым калием и хлористово- дородной кислотой). Положительные реакции на железо отличают гемосидерин от сходных с ним пигментов (гемомеланин, липофусцин, меланин).  Сидеробласт. Крупное ядро (Я), узкий ободок цитоплазмы с большим числом сидеросом (Сс). Электронограмма. х20 000 В условиях патологии наблюдается избыточное образование гемосидерина - гемосидероз. Он может иметь как общий, так и местный характер. Общий, или распространенный, гемосидероз наблюдается при внутрисосудистом разрушении эритроцитов (интраваскулярный гемолиз) и встречается при болезнях системы кроветворения (анемии, гемобластозы), интоксикациях гемолитическими ядами, некоторых инфекционных заболеваниях (возвратный тиф, бруцеллез, малярия и др.), переливаниях иногруппной крови, резус-конфликте и т.д. Разрушенные эритроциты, их обломки, гемоглобин идут на построение гемосидерина. Сидеробластами становятся ретикулярные, эндотелиальные и гистиоцитарные элементы селезенки, печени, костного мозга, лимфатических узлов, а также эпителиальные клетки печени, почек, легких, потовых и слюнных желез. Появляется большое число сидерофагов, которые не успевают поглощать гемосидерин, загружающий межклеточное вещество. В результате этого коллагеновые и эластические волокна пропитываются железом. При этом селезенка, печень, костный мозг и лимфатические узлы становятся ржаво-коричневыми. Местный гемосидероз - состояние, развивающееся при внесосудистом разрушении эритроцитов (экстраваскулярный гемолиз), т.е. в очагах кровоизлияний. Оказавшиеся вне сосудов эритроциты теряют гемоглобин и превращаются в бледные круглые тельца («тени» эритроцитов), свободный гемоглобин и обломки эритроцитов идут на построение пигмента. Сидеробластами и сидерофагами становятся лейкоциты, гистиоциты, ретикулярные клетки, эндотелий, эпителий. Сидерофаги могут долго сохраняться на месте бывшего кровоизлияния, нередко они переносятся током лимфы в близлежащие лимфатические узлы, где задерживаются, и узлы становятся ржавыми. Часть сидерофагов разрушается, пигмент высвобождается и в дальнейшем снова подвергается фагоцитозу. Гемосидерин образуется при всех кровоизлияниях, как мелких, так и крупных. В небольших кровоизлияниях, которые чаще имеют характер диапедезных, обнаруживается только гемосидерин. При крупных кровоизлияниях по периферии, среди живой ткани образуется гемосидерин, а в центре - кровоизлияния, где аутолиз происходит без доступа кислорода и участия клеток, появляются кристаллы гематоидина. В зависимости от условий развития местный гемосидероз может возникать в пределах не только участка ткани (гематома), но и целого органа. Таков гемосидероз легких, наблюдающийся при ревматическом митральном пороке сердца, кардиосклерозе и др. Хронический венозный застой в легких ведет к множественным диапедезным кровоизлияниям, в связи с чем в межальвеолярных перегородках, альвеолах, лимфатических сосудах и узлах легких появляется большое число на- груженных гемосидерином клеток (см. Венозное полнокровие).  Гемосидероз легких. Цитоплазма гистиоцитов и альвеолярного эпителия (сидеробластов и сидерофагов) нагружена зернами пигмента Близко к общему гемосидерозу своеобразное заболевание - гемохроматоз, который может быть первичным (наследственный гемохроматоз) или вторичным. Первичный гемохроматоз - самостоятельное заболевание из группы болезней накопления. Передается доминантно-аутосомным путем и связано с наследственным дефектом ферментов тонкой кишки, что ведет к повышенному всасыванию пищевого железа, которое в виде гемосидерина откладывается в большом количестве в органах. Обмен железа эритроцитов при этом не нарушен. Количество железа в организме увеличивается в десятки раз, достигая 50-60 г. Развивается гемосидероз печени, поджелудочной железы, эндокринных органов, сердца, слюнных и потовых желез, слизистой оболочки кишечника, сетчатки глаза и даже синовиальных оболочек; одновременно в органах увеличивается содержание ферритина. В коже и сетчатке глаз увеличивается содержание меланина, что связано с поражением эндокринной системы и нарушением регуляции меланинобразования. Основными симптомами болезни являются бронзовая окраска кожи, сахарный диабет (бронзовый диабет) и пигментный цирроз печени. Возможно развитие и пигментной кардиомиопатии с нарастающей сердечной недостаточностью. Вторичный гемохроматоз - заболевание, развивающееся при приобретенной недостаточности ферментных систем, обеспечивающих обмен пищевого железа, что ведет к распространенному гемосидерозу. Причиной этой недостаточности могут быть избыточное поступление железа с пищей (железосодержащие препараты), резекция желудка, хронический алкоголизм, повторные переливания крови, гемоглобинопатии (наследственные,заболевания, в основе которых лежит нарушение синтеза гема или глобина). При вторичном гемохроматозе содержание железа повышено не только в тканях, но и в сыворотке крови. Накопление гемосидерина и ферритина, наиболее выраженное в печени, поджелудочной железе и сердце, приводит к циррозу печени, сахарному диабету и кардиомиопатии. 12.Нарушения обмена билирубина, морфологическая характеристика. Желтухи. Билирубин - важнейший желчный пигмент. Его образование начинается в гистиоцитарно-макрофагальной системе при разрушении гемоглобина и отщеплении от него гема. Гем теряет железо и превращается в биливердин, при восстановлении которого образуется билирубин в комплексе с белком. Гепатоциты осуществляют захват пигмента, конъюгацию его с глюкуроновой кислотой и экскрецию в желчные капилляры. С желчью билирубин поступает в кишечник, где часть его всасывается и вновь попадает в печень, часть - выводится с калом в виде стеркобилина и мочой в виде уробилина. В норме билирубин встречается в растворенном состоянии в желчи и в небольшом количестве в плазме крови. Билирубин представлен красно- желтыми кристаллами. Он не содержит железа. Для выявления его употребляют реакции, основанные на способности пигмента легко окисляться с образованием различно окрашенных продуктов. Такова, например, реакция Гмелина, при которой под воздействием концентрированной азотной кислоты билирубин дает сначала зеленое, а затем синее или пурпурное окрашивание. Нарушение обмена билирубина связано с расстройством его образования и выделения. Это ведет к повышенному содержанию билирубина в плазме крови и желтому окрашиванию им кожи, склер, слизистых и серозных оболочек и внутренних органов - желтухе. Механизм развития желтухи различен, что позволяет выделять три ее вида: надпеченочную (гемолитическую), печеночную (паренхиматозную) и подпеченочную (механическую). Надпеченочная (гемолитическая) желтуха характеризуется повышенным образованием билирубина в связи с увеличенным распадом эритроцитов. Печень в этих условиях образует большее, чем в норме, количество пигмента, однако вследствие недостаточности захвата билирубина гепатоцитами уровень его в крови остается повышенным. Гемолитическая желтуха наблюдается при инфекциях (сепсис, малярия, возвратный тиф) и интоксикациях (гемолитические яды), при изоиммунных (гемолитическая болезнь новорожденных, переливание несовместимой крови) и аутоиммунных (гемобластозы, системные заболевания соединительной ткани) конфликтах. Она может развиваться и при массивных кровоизлияниях, геморрагических инфарктах в связи с избыточным поступлением билирубина в кровь из очага распада эритроцитов, где желчный пигмент выявляется в виде кристаллов. С образованием в гематомах билирубина связано изменение их окраски. Гемолитическая желтуха может быть обусловлена дефектом эритроцитов. Таковы наследственные ферментопатии (микросфероцитоз, овалоцитоз), гемоглобинопатии, или гемоглобинозы (талассемия, или гемоглобиноз F; серповидноклеточная анемия, или гемоглобиноз S), пароксизмальная ночная гемоглобинурия, так называемые шунтовые желтухи (при дефиците витамина В12, некоторых гипопластических анемиях и др.). Печеночная (паренхиматозная) желтуха возникает при поражении гепатоцитов, в результате чего нарушаются захват ими билирубина, конъюгация его с глюкуроновой кислотой и экскреция. Такая желтуха наблюдается при остром и хроническом гепатитах, циррозах печени, медикаментозных ее повреждениях и аутоинтоксикации, например при беременности, ведущих к внутрипеченочному холестазу. Особую группу составляют ферментопатические печеночные желтухи, возникающие при наследственных пигментных гепатозах, при которых нарушена одна из фаз внутрипеченочного обмена билирубина. Подпеченочная (механическая) желтуха связана с нарушением проходимости желчных протоков, что затрудняет экскрецию и определяет регургитацию желчи. Эта желтуха развивается при наличии препятствий оттоку желчи из печени, лежащих внутри или вне желчных протоков, что наблюдается при желчнокаменной болезни, раке желчных путей, головки поджелудочной железы и сосочка двенадцатиперстной кишки, атрезии (гипоплазии) желчных путей, метастазах рака в перипортальные лимфатические узлы и печень. При застое желчи в печени возникают очаги некроза с последующим замещением их соединительной тканью и развитием цирроза (вторичный билиарный цирроз). Застой желчи приводит к расширению желчных протоков и разрыву желчных капилляров. Развивается холемия, которая вызывает не только интенсивную окраску кожи, но и явления общей интоксикации, главным образом от воздействия на организм циркулирующих в крови желчных кислот (холалемия). В связи с интоксикацией понижается способность крови к свертыванию, появляются множественные кровоизлияния (геморрагический синдром). С аутоинтоксикацией связано поражение почек, развитие печеночно-почечной недостаточности. 13.Кальцинозы Виды, причины, патогенез, морфогенез, клинико-морфологические проявления, исходы. |