Генетика. 34. Ген как структурная и функциональная единица наследственности. Классификация и свойства генов. Регуляторные гены и регуляторные последовательности
 Скачать 3.46 Mb. Скачать 3.46 Mb.
|

|
60. Основные положения хромосомной теории. Сцепленное наследование. Типы генетических карт. Хромосомная теория наследственности была сформулирована Т. Морганом, основные положения которой сводятся к следующему: • гены находятся в хромосомах, каждый ген занимает в хромосоме определенное место (локус); • гены в хромосомах располагаются линейно; • каждая хромосома представляет собой группу сцепления генов; • число групп сцепления у каждого вида равно гаплоидному набору хромосом; • между гомологичными хромосомами в процессе кроссинговера происходит обмен аллельными генами, что приводит к формированию новых сочетаний аллелей в группах сцепления; • расстояние между генами в хромосоме пропорционально проценту кроссинговера между ними. Изучение сцепленного наследования явилось основой для составления генетических карт сцепления у разных организмов. Методы классической генетики, цитогенетики и молекулярной генетики позволили подойти к составлению современных генетических карт Генетическая карта — это система элементов генома, упорядоченная на основе хромосомной принадлежности и взаимного расположения генов в пределах отдельных хромосом, т. е. она определяет принадлежность генов к хромосоме и их расположение относительно друг друга. Генетическое картирование необходимо для определения нуклеотидной последовательности гена и прилегающих к нему участков. Выделяют следующие генетические карты: карты сцепления, цитологические карты, цитогенетические карты индивидуальных хромосом, рестрикционные и секвенсовые карты. Карта сцепления — схема расположения генов, находящихся в одной группе сцепления, т. е в одной хромосоме. Цитологическая карта составляется на основании изучения политенных хромосом, что позволяет сопоставить структуру синтезируемого белка с определенным участком хромосомы (геном), так как транскрибируемый участок определяется под микроскопом в виде пуфа. Цитогенетические карты хромосом составляются на основе дифференциальной окраски (темные и светлые полосы) и картирования генов в отдельных локусах хромосом (основа Парижской классификации). Генетическое картирование необходимо для определения нуклеотидной последовательности гена и прилегающих к нему участков. Рестрикционные карты ДНК представляют собой участки ДНК с определенной нуклеотидной последовательностью. Секвенсовые карты содержат данные о последовательности всех нуклеотидов в целой молекуле ДНК, а не в отдельных ее фрагментах. 61. Наследственные болезни человека. Классификация. Примеры. Понятие о мультифакториальных болезнях. Примеры. Наследственные болезни - это структурные или функциональные патологические изменения организма, вызванные изменением наследственной информации. В настоящее время известно более двух тысяч наследственных болезней. Согласно одной из классификаций, их можно разделить на генные, хромосомные и мультифакториальные (болезни с наследственным предрасположением) Мультифакториальные болезни (см.вопр. №63) Развитие некоторых болезней (гипертоническая болезнь, атеросклероз, язвенная болезнь, определенные формы диабета, шизофрения и др.) зависит как от генотипа (полигенное наследование), так и от внешней среды. Эти болезни называются болезнями с наследственным предрасположением. Для них характерно изменение нормы реакции на действие факторов внешней среды (при предрасположенности к диабету изменяется норма реакции на крахмал и сахар - увеличение глюкозы в крови). Для мультифакториальных болезней характерно: - отсутствие типичного расщепления, как при моногенном наследовании; - различие в частоте заболеваний у родственников первой и второй степени родства; -широкое варьирование количественных признаков (размах изменчивости) среди населения; - зависимость проявления заболеваний от возраста, пола, питания и др.; - отсутствие резких различий в проявлении признаков между здоровыми и больными людьми. 62. Хромосомные болезни, причины и механизмы их возникновения. Классификация. Примеры. Хромосомные болезни возникают при нарушении кариотипа (нормальный кариотип человека 46,ХХ, 46,ХУ), которое может быть вызвано изменением числа (геномные мутации) или структуры (хромосомные мутации) аутосом и половых хромосом. При хромосомных болезнях имеется определенный комплекс стабильных аномальных признаков - симптомов, который входит в понятие синдром. Синдромы характеризуются определенной частотой проявления, продолжительностью жизни детей и средним весом при рождении, внешними морфологическими признаками, пороками развития внутренних органов, функциональными симптомами, дерматоглификой и определенным кариотипом. Среди хромосомных болезней, связанных с изменением числа половых хромосом (моносомии, полисомии), наиболее часто встречаются синдромы Клайнфельтера (47,ХУ), Шерешевского-Тернера (45,Х), трисомия по Х-хромосоме (47,ХХХ), полисомия по У-хромосоме (47,ХУУ); может наблюдаться полисомия по Х-хромосоме и У-хромосоме одновременно ( 48,ХХУУ). Эффект мутаций аутосом различен: при геномных мутациях 1-12 хромосом возникают аномалии, несовместимые с жизнью; 13-18 - полулетальные мутации (спонтанные аборты, множественные уродства, незначительная продолжительность жизни родившихся от нескольких недель до нескольких лет - синдром Патау (47,ХХ/ХУ + 13), синдром Эдвардса (47,ХУ/ХХ + 18). Трисомия по 21 хромосоме - синдром Дауна (47,ХХ/ХУ + 21) - является аномалией, совместимой с жизнь. При анализе синдромов по половым хромосомам и аутосомам показано, что при аномалиях по половым хромосомам сохраняется нормальный интеллект или отмечается его снижение, но в большей степени нарушается развитие половых органов и гормонозависимый рост (выше или ниже средней нормы). Моносомия Х встречается реже (1: 25ОО), чем полисомии ХХУ (1: 700) и ХХХ (1: 1000). Хромосомные аберрации в основном представлены делециями и транслокациями: при делеции короткого плеча 5 хромосомы (46,ХХ/ХУ 5р-) наблюдается синдром «кошачьего крика» (название обусловлено сходством плача ребенка с мяуканьем кошки), происхождение которого вызвано нарушением центральной нервной системы, а не аномалией голосового аппарата. Встречаются делеции по 13, 18, 21, 22 хромосомам (46,ХХ/ХУ 13q- - синдром Орбели). Транслокация 15/21 хромосом приводит к возникновению синдрома Дауна; а 9/22- хроническому миелолейкозу. Для диагностики хромосомных болезней используется цитогенетический метод (кариотипирование, определение полового хроматина); методы амниоцентеза и дерматоглифики. Хромосомные болезни не наследуются, так как у больных нарушена репродуктивная функция, но синдромы появляются в каждом поколении с определенной частотой как результат вновь возникших мутаций у здоровых людей. 63. Молекулярные болезни, причины и механизмы их развития. Многообразие молекулярной патологии. Типы наследования молекулярных болезней. Генные болезни (молекулярные болезни) Генные болезни являются результатом генных мутаций: изменение структуры молекулы ДНК изменение РНК изменение полипептидной цепи изменение белка (структурного или белка фермента). Мутации возникают в аутосомах и половых хромосомах, они могут быть доминантными и рецессивными. По доминантному типу наследуются болезни, связанные с нарушением структурных, транспортных белков, белков-рецепторов и др. - это болезни костной системы и соединительной ткани (ахондроплазия, синдром Марфана, нейрофиброматоз и др.). Для диагностики этих болезней иногда используют и данные электронной микроскопии (изменение мышечных волокон, клеток эпидермиса, хондроцитов). По рецессивному типу (аутосомному или сцепленному с полом) наследуются многие болезни обмена веществ - энзимопатии или ферментопатии. Они возникают в результате мутации генов, ответственных за синтез ферментов (Д. Бидл, Э.Тейтум). У человека большинство ферментопатий вызвано мутациями структурных генов, что приводит к качественному, а не количественному изменению ферментов. Наиболее распространены энзимопатии, обусловленные нарушением аминокислотного обмена (фенилкетонурия), жирового (амавротическая идиотия), углеводного (галактоземия, фруктозурия ), пуринового и пиримидинового (синдром Леш-Нихана), минерального (нарушение обмена меди - болезнь Вильсона-Коновалова). 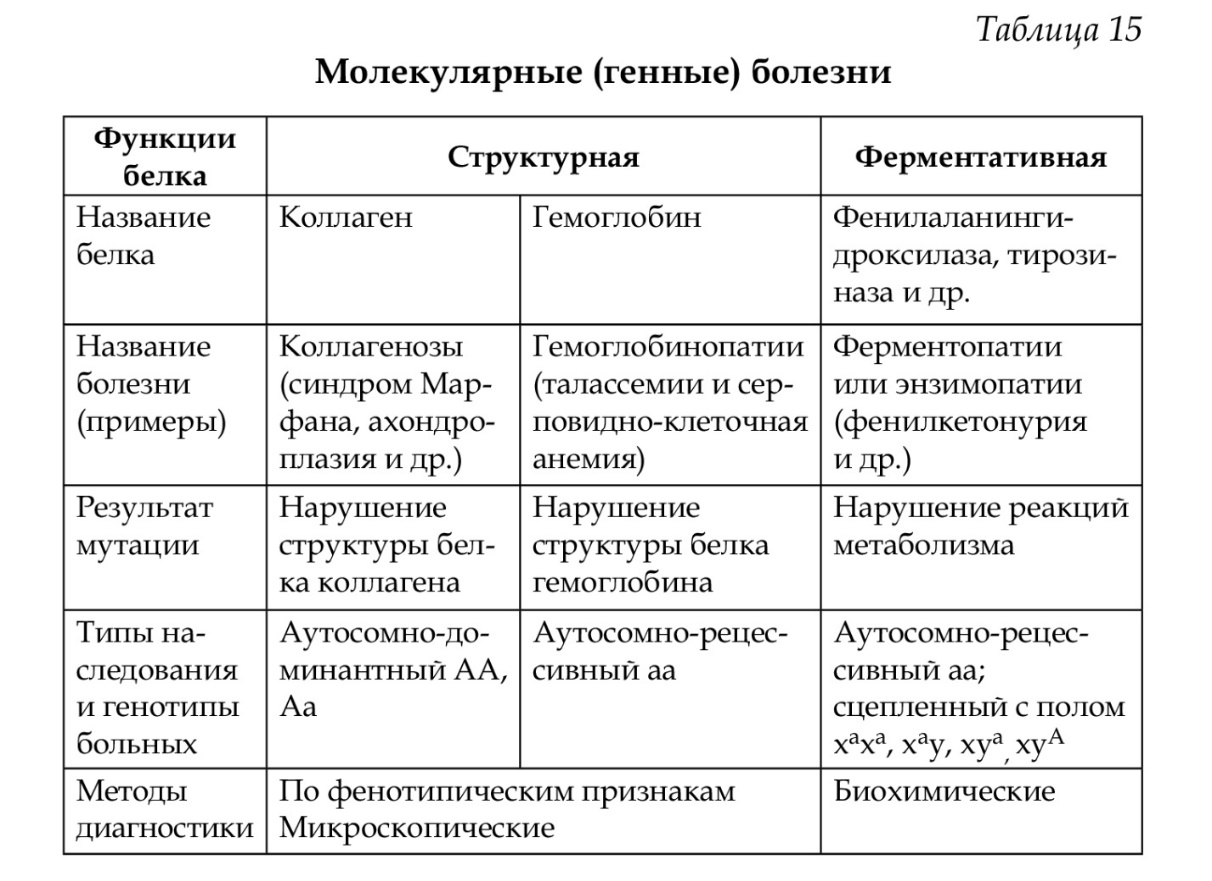 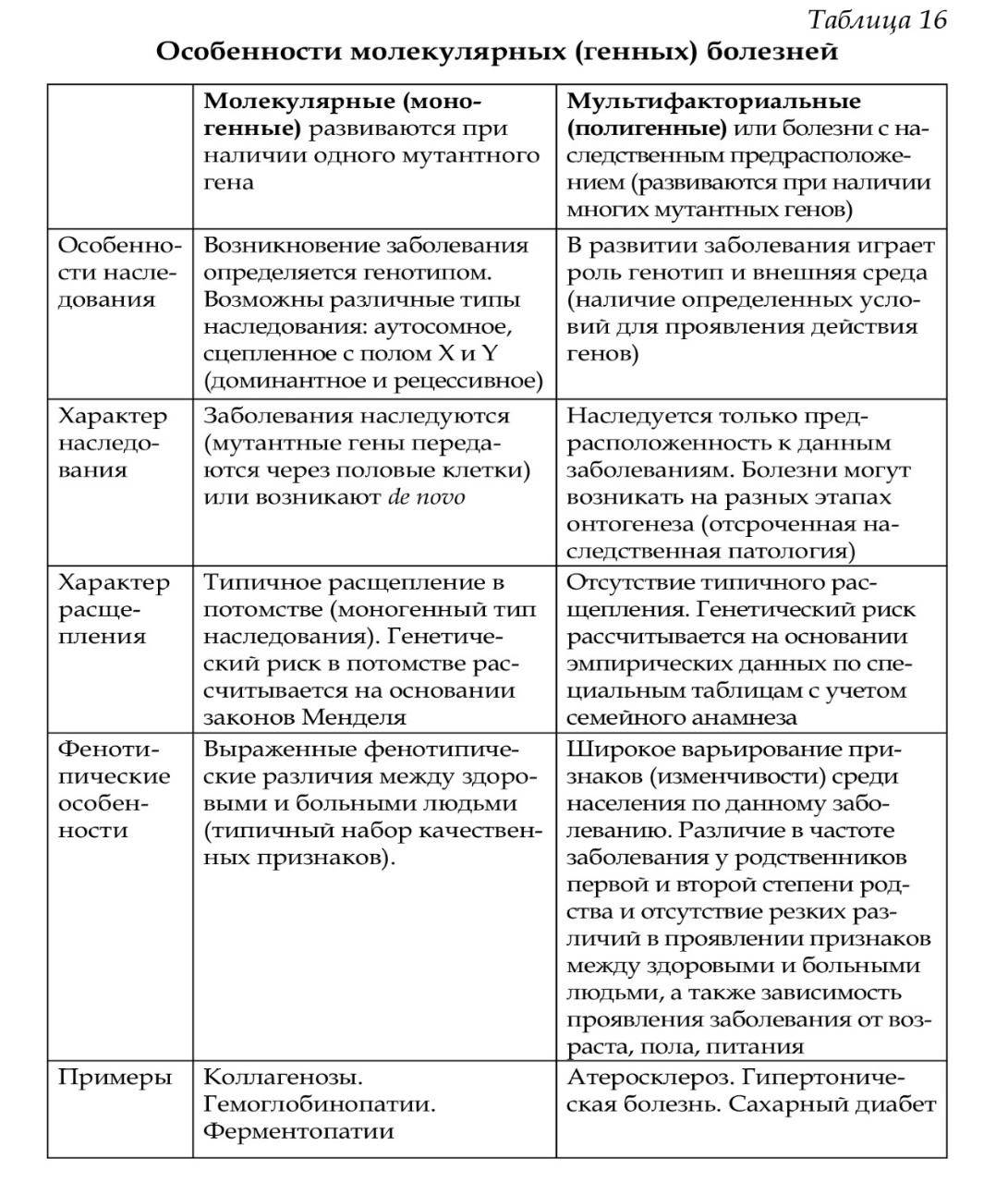 64. Ферментопатии. Классификация, механизмы возникновения, типы наследования. Примеры. Моделью для изучения энзимопатий может служить фенилкетонурия. Классическая фенилкетонурия вызвана мутацией гена РАН (пи эй эйч), он картирован (12q 22), идентифицирован и секвенирован (определена последовательность нуклеотидов). Ген РАН относится к мозаичным генам и состоит из 13 экзонов и 12 интронов; он детерминирует синтез фермента фенилаланингидроксилазы — ФАГ. Заболевание связано с нарушением обмена аминокислоты фенилаланина. В норме аминокислота фенилаланин превращается в аминокислоту тирозин, а тирозин — в пигмент меланин. Мутация гена вызывает уменьшение активности фермента ФАГ, в результате этого фенилаланин не полностью превращается в тирозин. Фенилаланин накапливается в крови и частично превращается в фенилпировиноградную кислоту (ФПК), которая выделяется с мочой и потом — от больных исходит «мышиный запах». ФПК является нейротропным ядом (нарушается формирование миелиновой оболочки вокруг аксонов ЦНС), поэтому у детей развивается повышенная возбудимость, тремор, судорожные эпилептиформные припадки, происходит нарушение высшей нервной деятельности, развивается тяжелая умственная отсталость. Диагностика фенилкетонурии осуществляется биохимическими (определение фенилаланина в крови и ФПК в моче), микробиологическими (тест Гартри), молекулярно-генетическими и клиническим методами. Предупреждение тяжелых последствий в развитии болезни базируется на диетотерапии: используются белковые гидролизаты с уменьшенным количеством фенилаланина и специаль-ные наборы продуктов (мед, орехи и др.). Лечение детей проводится до 7-10 лет, мозг взрослого человека устойчив к высоким концентрациям ФПК. При нарушении активности фермента тирозиназы (мутация гена) не происходит превращение тирозина в меланин и возникает альбинизм. У больных наблюдаются слабая пигментация кожи, волос, радужной оболочки, изменения в почках, печени, селезенке. В ряде случаев развивается алкаптонурия, причиной возникновения которой является генетический дефект фермента оксидазы; в результате этого гомогентизиновая кислота (промежуточный продукт обмена) не расщепляется полностью до воды и углекислого газа, что происходит в норме, а откладывается в соединительной ткани (цвет охры) и выводится с мочой (темная моча). Данные заболевания наследуются по аутосомно-рецессивному типу, встречаются с относительно высокой частотой 1 : 10000, при частоте мутантного гена у гетерозигот в человеческих популяциях 1 : 50 (1 : 75). Профилактика: медико-генетическое консультирование на разных этапах онтогенеза, исключение браков между гетерозиготами, а для этого выявление гетерозигот с использованием соответствующих методов. В настоящее время проводится скрининг всех новорожденных на фенилкетонурию, гипотиреоз и муковисцидоз.  65. Методы диагностики молекулярных и хромосомных болезней. Пренатальная диагностика наследственных болезней человека. Причиной возникновения наследственной патологии являются мутации (вновь возникающие или унаследованные от предыдущих поколений), поэтому уменьшение числа мутаций в популяциях является одной из важнейших задач. Профилактика может быть первичной (предупреждение зачатия или рождения больного ребенка) и вторичной (разнообразные способы коррекции проявления патологического генотипа). Профилактика наследственных заболеваний может проводиться на разных этапах онтогенеза человека: преэмбриональном, эмбриональном, постэмбриональном. Преэмбриональный период включает гаметогенез и оплодотворение, эмбриональный период начинается от момента образования зиготы и продолжается до рождения ребенка, постэмбриональный период — от момента рождения до окончания жизни. 1. Преэмбриональный: — мероприятия по улучшению среды обитания и уменьшению контакта с мутагенами: физическими (рентгеновское облучение и др.), химическими (лекарственные препараты, алкоголь, никотин, наркотики и др.), биологическими (например, вирусные заболевания — краснуха, СПИД и др.); — применение антимутагенов (витамины, аминокислоты, радио-протекторы и др.); — химический скрининг с использованием специальных тест-систем (контроль за содержанием мутагенов в продуктах питания, лекарствах и др.); — планирование семьи (см. выше); — медико-генетическое консультирование (см. выше); — пропаганда здорового образа жизни (вред алкоголя, никотина, наркотиков). 2. Эмбриональный. При подозрении на наследственные болезни и с целью их ранней диагностики: — определение генотипа и кариотипа плода на ранних сроках беременности с использованием инвазивных и неинвазивных методов; — обследование беременной женщины (цитогенетический анализ, определение альфа-фетопротеина и хорионического гонадотропина, как маркеров некоторых наследственных забо-леваний). 3. Постэмбриональный: — массовое скринирование новорожденных на наследственные болезни с использованием скрининг-тестов (скрининг-диагностика). Скрининг-диагностика — бесплатное тотальное обследование всех новорожденных на фенилкетонурию, врожденный гипотиреоз, муковисцидоз. Применяется для ди-агностики заболеваний, которые имеют высокую распростра-ненность в популяциях и приводят к ранней инвалидизации. При раннем выявлении и лечении симптомы заболеваний не проявляются. Профилактика возникновения наследственных заболеваний проводится как на уровне отдельных семей, так и на уровне популяций 66. Понятие о резусе-факторе, закономерности наследования, профилактика резус-конфликта. В Европе у 85% людей на поверхности эритроцитов имеется антиген (белок), который называется резус-фактор (они условно называются резус-положительными), у 15% — в эритроцитах такого антигена нет (они условно называются резус-отрицательными). Резус-фактор наследуется по доминантному типу (наличие резус-фактора RhRh, Rhrh, его отсутствие — rhrh). Иногда при переливании крови и некоторых вариантах браков возникает резус-несовместимость. Классическим примером несовместимости по резус-фактору матери и плода является вариант, когда в организме резус-отрицательной матери развивается резус-положительный плод. Кровь матери и плода разделена (плацента — биологическая мембрана), эритроциты через мембрану не проникают. Во время родов эритроциты плода, имеющие антиген, могут попасть в кровоток матери и вызвать образование антител. Антитела свободно перемещаются через плаценту из организма матери в организм плода. Эритроциты второго ребенка с момента начала кроветворения в эмбриогенезе уже будут подвергаться воздействию образовавшихся антител матери (реакция антитело-антиген на ПАК эритроцитов) и разрушаться (гемолиз эритроцитов). В результате гемолиза эритроцитов возникает гемолитическая болезнь, симптомами которой являются анемия, желтуха, водянка. Для спасения ребенка после рождения ему осуществляют обменное переливание одногрупповой крови, при котором восстанавливается дыхательная функция крови и удаляются токсические продукты. Для профилактики гемолитической болезни у последующих детей женщине до родов или сразу после родов вводят антирезусные антитела, которые связывают антигены (эритроциты плода), попавшие в кровь (эффект Кларка — профилактика резус-несовместимости). Иммунная система матери остается интактной. 67. Генетика пола, механизм генетической детерминации и дифференцировки пола в онтогенезе. Пол - это совокупность морфологических, физиологических, биохимичеких, поведенческих и других признаков организма, обусловливающих репродукцию. Определение пола происходит в момент оплодотворения и зависит от набора половых хромосом в зиготе (46,ХХ/46,ХУ). В дальнейшем наблюдается дифференцировка пола: образование гонад (семенников и яичников) - формируется гонадный пол. Гонады вырабатывают гормоны, в клетках-мишенях происходит взаимодействие молекул гормона с белками рецепторами - формируется гормональный пол, что обеспечивает в последующем формирование вторичных половых признаков по мужскому или женскому типу (акушерский или фенотипический пол). В дифференцировке мужского пола участвует андроген (находится в У-хромосоме), который детерминирует образование НУ- антигена, а он, в свою очередь, определяет развитие семенников. Наряду с андрогеном, в этом участвут ген tfm, который определяет образование в цитоплазме клеток-мишеней белков-рецепторов, взаимодействующих с тестостероном. При мутации гена tfm наблюдается синдром тестикулярной феминизации (синдром Морриса) - женский фенотип при мужском кариотипе. Для нормального развития пола необходимо наличие двух половых хромосом (ХХ или ХУ). При геномных мутациях (хромосомные болезни) наблюдается их увеличение или уменьшение, что приводит к недоразвитию первичных и вторичных половых признаков. 68. Медико-генетическое консультирование, его этапы и роль в профилактике, диагностике и лечении наследственной патологии человека. Примеры. Профилактика возникновения наследственных болезней проводится по нескольким направлениям: - на уровне популяций - уменьшение действия мутагенных факторов, применение антимутагенов, разработка и использование специальных скрининг-программ на наследственные болезни (диагностика и профилактика болезней обмена, хромосомных болезней); - на уровне семей (медико-генетическое консультирование). Медико-генетическое консультирование складывается из нескольких этапов: I. Диагностика заболевания: - установление природы наследственного заболевания (генное, хромосомное); - обследование больного с применением соответствующих методов изучения генетики человека (генеалогического, цитогенетического, биохимического и др.); - выяснение уровня нарушения наследственного материала: на уровне молекулы ДНК, хромосом, генома; - при генных болезнях - установление связи между патологией и изменением белка (структурного или белка-фермента); - при хромосомных болезнях - установление связи между патологией и типом хромосом (аутосомы или половые хромосомы); - сопоставление полученных данных у обследуемого больного с фенотипической картиной известных наследственных болезней. II. Определение вероятности развития болезни в потомстве при моногенном (аутосомно-доминантное, аутосомно-рецессивное и сцепленное с полом) и полигенном типе наследования. III. Заключение: рекомендации врача-генетика родителям (сопоставление риска рождения ребенка с тяжестью заболевания и возможными социальными последствиями). Определение генетического риска складывается из 3 моментов: вероятность появления данной аномалии (в процентах от 1 до 100 при аутосомно-доминантном и аутосомно-рецессивном, сцепленном с полом наследовании); оценка тяжести медицинских и социальных последствий; перспектива применения и эффективность методов пренатальной диагностики. Медико-генетическое консультирование может проводиться при обследовании вступающих в брак, обследование матери во время беременности (пренатальная диагностика эмбриона или плода), обследование ребенка после рождения. 69. Методы молекулярной генетики. Генная инженерия, ее значение для медицины, перспективы развития. Значение генетики для медицины. Знание структуры и функции генов, основных видов изменчивости, знакомство с наследственными болезнями позволяет перейти к анализу молекулярно-генетических методов. Методы молекулярной генетики направлены на изучение молекулы ДНК как в норме, так и при ее повреждении, а также на «манипуляции» с молекулами ДНК и РНК. Использование молекулярно-генетических методов требует знания основных этапов получения определенных последовательностей (фрагментов) ДНК. Этапы получения последовательностей нуклеотидов ДНК: 1. Получение образцов ДНК: — выделение всей (геномной) ДНК из — рестрикция ДНК — получение отдельных фрагментов ДНК 2. Амплификация — накопление (умножение, клонирование) одинаковых фрагментов ДНК 3. Электрофорез фрагментов ДНК — разделение фрагментов по молекулярной массе и электрическому заряду на поверхности агарозного геля. 4. Идентификация конкретных фрагментов ДНК с помощью блот-гибридизации по Саузерну. Базируясь на технологии получения нуклеотидных последовательностей ДНК, можно выделить следующие методы молекулярной генетики. 1. Метод секвенирования — определение нуклеотидной последовательности ДНК. Таким методом полностью определена последовательность нуклеотидов генов глобина, некоторых гормонов (инсулина, гормона роста, пролактина и др.). 2. Метод полимеразной цепной реакции (используется для увеличения числа фрагментов ДНК). 3. Метод получения праймеров, соответствующих известным генам. 4. Метод гибридизации нуклеиновых кислот. 5. Метод клонирования ДНК. 6. Метод получения рекомбинантных молекул ДНК. 7. Метод получения белков с помощью рекомбинантных молекул ДНК. 8. Создание библиотеки генов — полного набора (коллекции) клонированных фрагментов ДНК, полученных в результате рестрикции тотальной ДНК. Методы молекулярной генетики позволяют: • идентифицировать мутации в гене. Примером выявления мутантного гена является диагностика серповидно-клеточной анемии в эмбриональном периоде. Фрагменты ДНК, полученные при действии рестриктаз у здорового и больного, сравниваются с помощью метода гибридизации по Саузерну, при этом в качестве зонда используется радиоактивно меченая ДНК гена Р-глобина; • диагностировать моногенное наследственное заболевание путем определения нуклеотидной последовательности генов (гемофилия, гемоглобинопатия) и выявления мутантных генов (фенилкетонурия, муковисцидоз); • осуществлять генетический анализ полиморфизма ДНК родителей и детей; • определять индивидуальную изменчивость ДНК человека по вариабельным точкам ДНК, молекулярный анализ которых по-зволяет проводить идентификацию личности человека); • выделять и синтезировать гены (выделение, синтез и кло-нирование генов является одним из этапов генной инженерии). |