анализ поликарбоновых кислот по гф 14. курсовая работа. Анализ поликарбоновых кислот по гф 14
 Скачать 1.85 Mb. Скачать 1.85 Mb.
|

|
Хроматографические условия Оксалаты. Не более 0,35 %. Определение проводят методом спектрофотометрии. Суть этого метода сводится к следующему: салицилат железа имеет сине- или красно-фиолетовое окрашивает. Оксалат железа сам по себе бесцветен, поэтому при добавлении щавелевой кислоты или её соли к салицилату железа окраска последнего теряет свою интенсивность, что будет заметно при снятии показаний спектрофотометра Раствор железа салицилата. 0,1 г железа(III) аммония сульфата растворяют в 100 мл 2 % (о/о) раствора серной кислоты. К полученному раствору прибавляют 50 мл 1,15 % раствора натрия салицилата, 10 мл 2 М раствора уксусной кислоты, 80 мл 13,6 % раствора натрия ацетата и доводят объем раствора водой до 500 мл. Испытуемый раствор. 0,1 г субстанции растворяют в 20 мл воды, прибавляют 5 мл раствора железа салицилата и доводят объем раствора водой до 50,0 мл. Стандартный раствор. 0,35 г щавелевой кислоты растворяют в 20 мл воды, прибавляют 5 мл раствора железа салицилата и доводят объем раствора водой до 50,0 мл. Оптическая плотность испытуемого раствора, измеренная на спектрофотометре при длине волны 480 нм в кювете с толщиной слоя 10 мм относительно воды, не должна превышать оптическую плотность стандартного раствора, измеренную в тех же условиях. Количественное определение Определение проводят методом титриметрии Около 0.2 г субстанции растворяют при нагревании в смеси 5 мл пропан-2-ола и 25 мл этиленгликоля и охлаждают. Прибавляют 30 мл диоксана и титруют 0.1М раствором хлорной кислоты. КТТ определяют потенциометрически Параллельно проводят контрольный опыт 1 мл 0.1М хлорной кислоты соответствует 25.62 мг кромогликата натрия 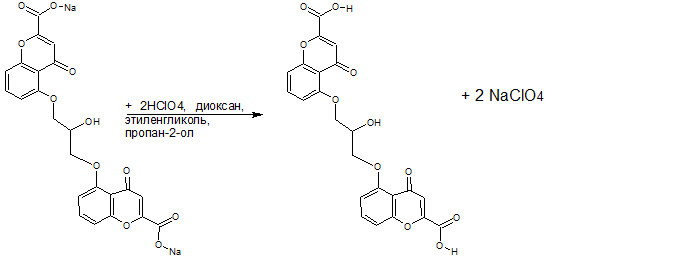 Теперь мы рассмотрим отдельно следующие соли Фумараты Сукцинаты Тартраты/гидротартраты Оксалаты Малеаты И, собственно, их кислоты, возможные методы их качественного и количественного анализа Фумаровая кислота (ФК)/ фумараты Химическое наименование – (2Е)-Бут-2-ен-1,4-диовая кислота Описание Ф 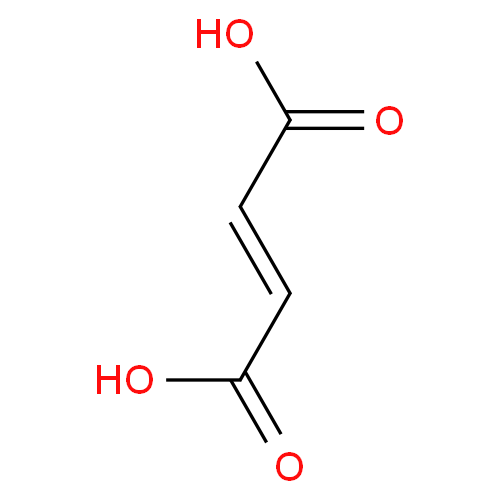 умаровая кислота представляет собой бесцветное кристаллическое твердое вещество - иглы, моноклинные призмы или белый кристаллический порошок без запаха умаровая кислота представляет собой бесцветное кристаллическое твердое вещество - иглы, моноклинные призмы или белый кристаллический порошок без запахаРастворимость Мало растворима в воде - 7,0 мг / мл, растворим в этаноле, концентрированной серной кислоте; мало растворим в этиловом эфире, ацетоне pKa1 = 3,03; pKa2 = 4,54 при 25 ° C Итак, каковы же методы ее анализа? Прежде всего давайте обратимся к фармакопеям – ГФ 14 издания и европейской фармакопее 8 издания – и выясним на примере каких субстанций мы можем анализировать ФК ЛП, содержащие ФК в ГФ 14 Б  исопролола фумарат Клемастина фумарат исопролола фумарат Клемастина фумарат ЛП, содержащие ФК в ЕФ 8 издания 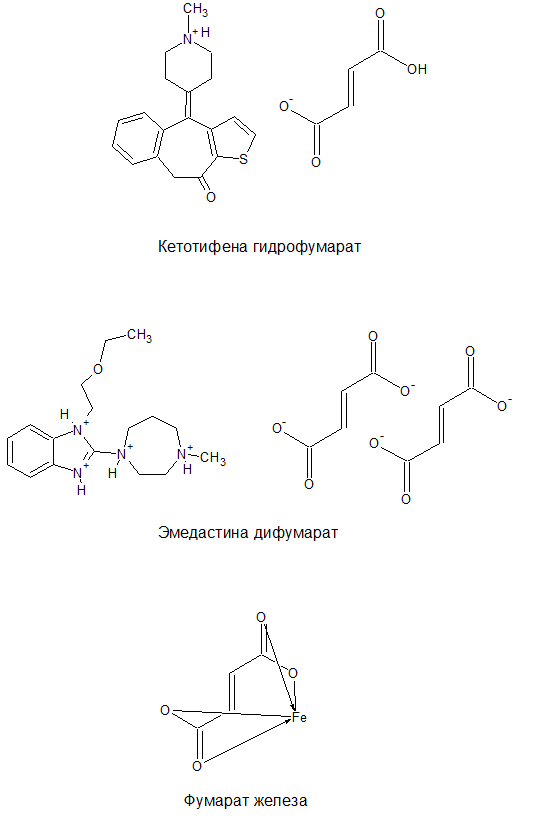 Общими методами анализа для всех выше перечисленных субстанций являются различные хроматографические методы – например, ВЭЖХ, ТСХ – и ИК-спектрометрия, в отдельных случаях УФ-спектрофотометрия * *Здесь необходимо сделать пометку, что УФ-СФ применяется в основном ввиду особенностей поглощения не фумаровой кислоты, а катионной составляющей соли ФК – так, например, максимум поглощения бисопролола должен быть при 222 ±2 нм, который обеспечен именно за счет бисопролола, а не ФК – УФ-спектр ФК будет рассмотрен позднее. Примеры анализа ИК-спектроскопия.(на примере бисопролола фумарата) Инфракрасный спектр субстанции, снятый в диске с калия бромидом, в области частот от 4000 до 400 см-1 по положению полос поглощения должен соответствовать спектру стандартногообразца бисопролола фумарата. Если спектры различаются, испытуемую субстанцию и стандартный образец бисопролола фумарата растворяют в метаноле, выпаривают досуха, сушат при остаточном давлении 0,7 кРа и температуре 60 ºС и снимают спектры сухих остатков. ВЭЖХ (на примере бисопролола фумарата и эмедастина дифумарата) Бисопролола фумарат Растворитель. Вода – ацетонитрил (65:35). Подвижная фаза: К 5,0 мл гептафтормасляной кислоты прибавляют 5,0 мл диэтиламина и 2,5 мл муравьиной кислоты. Доводят объём растворителем до 1000 мл. Испытуемый раствор. Около 50 мг (точная навеска) субстанции растворяют в растворителе и доводят объём растворителем до 50,0 мл. Раствор сравнения. Около 20 мг (точная навеска) стандартного образца бисопролола фумарата растворяют в растворителе и доводят объём растворителем до 20,0 мл. Раствор для проверки пригодности хроматографической системы. 5 мг пропранолола гидрохлорида растворяют в 10 мл раствора сравнения. Хроматографические условия Колонка - 12,5 × 0,46 см с октадецилсилилсиликагелем (С18), 5 мкм; Температура колонки - 30 0С Скорость потока - 1,0 мл/мин; Детектор - спектрофотометрический, 273 нм; (!) Объем пробы - 10 мкл. Пригодность системы: - на хроматограмме раствора для проверки пригодности хроматографической системы разрешение (R) между пиками бисопролола и пропранолола должно быть не менее 7,0; - на хроматограмме раствора сравнения фактор ассиметрии пика бисопролола должен быть не более 2,0; - на хроматограмме раствора сравнения относительное стандартное отклонение площади пика бисопролола не должно превышать 2,0 % (6 определений). Допустимое содержание примесей. На хроматограмме испытуемогораствора отношение суммарной площади пиков, кроме фумаровой кислоты ибисопролола, к суммарной площади всех пиков должно составлять не более0,5 %. (!) – хочется отметить, что пик УФ-поглощения для фумаровой кислоты составляет 209 нм, но детектор используют при 273 нм – т.е. даже при этой величине можно задетектировать ФК в субстанции 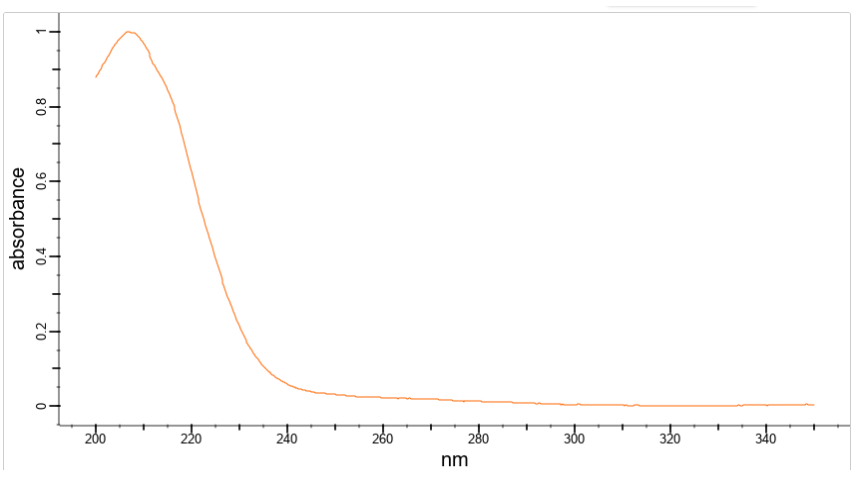 Уф-спектр поглощения фумаровой кислоты Эмедастина дифумарат Испытуемый раствор: растворите 10 мг исследуемого вещества в подвижной фазе и разбавьте подвижной фазой до 10 мл. Эталонный раствор (а). Растворите 5 мг примеси E CRS эмедастина в подвижной фазе и разбавьте подвижной фазой до 25 мл. Эталонный раствор (б). Растворите 10 мг исследуемого вещества в подвижной фазе. Добавьте 0,5 мл раствора сравнения (а) и разбавьте подвижной фазой до 10 мл. Эталонный раствор (с). Разбавьте 5,0 мл тестового раствора до 50,0 мл подвижной фазой. Разбавьте 1,0 мл этого раствора до 100,0 мл подвижной фазой. Колонка: - размер: l = 0,15 м, Ø = 4,6 мм; Неподвижная фаза: октадецилсилилсиликагель для хроматографии (5 мкм). Подвижная фаза: растворяют 3,9 г гидрофосфата динатрия и 2,5 г додецилсульфата натрия в воде и разбавляют тем же растворителем до 1000,0 мл. Довести pH до 2,4 с помощью фосфорной кислоты. Смешать 550 объемов этого раствора с 450 объемами ацетонитрила. Скорость потока: 1,0 мл / мин. Детектирование: спектрофотометр при 280 нм. (!) Для инъекций: 10 мкл исследуемого раствора и растворов сравнения (b) и (c). Время работы: вдвое больше времени удерживания эмедастина. Относительное удерживание относительно эмедастина (время удерживания = около 18 мин): фумаровая кислота = около 0,1; примесь А = около 0,2; примесь В = около 0,3; примесь C = около 0,5; примесь D = около 0,7; примесь E = около 0,9; примесь F = около 1,4. Тонкослойная хроматография по ГФ 14 (А) и ЕФ 8 (Б) А Пластинка. ТСХ пластинка со слоем силикагеля G. Подвижная фаза (ПФ). Вода – муравьиная кислота – диизопропиловый эфир 4:20:56. Испытуемый раствор. 40 мг субстанции растворяют в 2,0 мл спирта 70 %. Раствор сравнения. 50 мг фумаровой кислоты растворяют в 10,0 мл спирта 96 %.На линию старта ТСХ пластинки наносят по 5 мкл испытуемого раствора (100 мкг) и раствора сравнения (25 мкг). Пластинку с нанесёнными пробами высушивают на воздухе, помещают в камеру с ПФ и хроматографируют восходящим способом. Когда фронт ПФ пройдет около 80 – 90 % длины пластинки от линии старта, ее вынимают из камеры, сушат до удаления следов растворителей, выдерживают в сушильном шкафу при температуре 100 – 105 °С в течение 30 мин и опрыскивают 1,6 % водным раствором калия перманганата. Б Испытуемый раствор Растворите 40 мг исследуемого вещества в метаноле и доведите до 10 мл тем же растворителем. Эталонный раствор. Растворите 11 мг CRS фумаровой кислоты в метаноле и разбавьте до 10 мл тем же растворителем. Пластина: целлюлоза для хроматографии F254 в качестве вещества покрытия. Подвижная фаза: вода R, безводная муравьиная кислота R, диизопропиловый эфир R (3: 7: 90). Наносят по 5 мкл испытуемого раствора, проведение ТСХ на этапе распространения фронта считается законченным, когда последний пройдет 80 – 90 % длины пластинки от линии старта Сушка: в токе теплого воздуха. Обнаружение: исследуют в ультрафиолетовом свете при 254 нм. Слегка опрыскайте 5 г / л раствора перманганата калия в 1,4% растворе серной кислоты. Осмотрите при дневном свете на прозрачность. Результаты: пятно, вызванное фумаровой кислотой, на хроматограмме исследуемого раствора по положению, цвету и интенсивности аналогично основному пятну на хроматограмме контрольного раствора. Для железа фумарата также в ЕФ 8 есть следующая реакция: Смешайте 0,5 г с 1 г резорцин. К 0,5 г смеси в тигель добавляют 0,15 мл раствора серной кислоты и осторожно нагревают.Образуется темно-красная полутвердая масса. К получившейся массе аккуратно добавляют воду, до 100 мл. Окраска раствора – оранжево-желтая, без опалесценции. Принцип данной реакции основан на образовании ауринового красителя – при нагревании с концентрированной серной кислотой фумаровая кислота разрушается с образованием муравьиной кислоты, которая, в свою очередь, взаимодействует с резорцином, вследствие чего и образуется ауриновый краситель. Данная реакция, насколько это разумно предположить, подходит для субстанций, доля фумаровой кислоты достаточно высока, иначе эффект реакции нельзя будет заметить (т.е. можно предположить, что воспроизводимость данной реакции для других субстанций ФК - либо отсутствует, либо очень мала) Количественное определение фумаровой кислоты проводят титриметрическим методом Около 0.5 г (точная навеска) субстанции растворяют в 70 мл этанола (3484), прибавляют 8 мл 0.1 М раствора тетрабутиламмония гидроксида, перемешивают в течение 2 минут и титруют 0.1М раствором тетрабутиламмония гидроксида. КТТ определяют потенциометрически. (ОФС «Потенциометрическое титрование») С КО 1 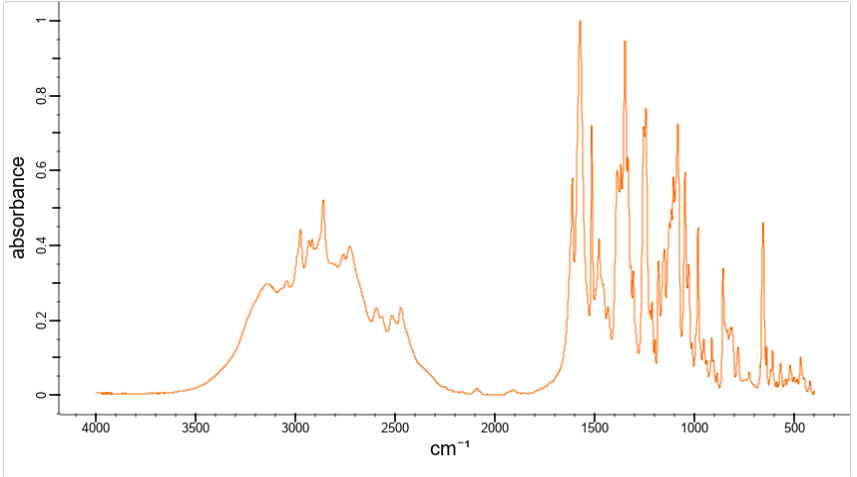 мл 0.1М раствора тетрабутиламмония гидроксида соответствует 5,804 мг фумаровой кислоты мл 0.1М раствора тетрабутиламмония гидроксида соответствует 5,804 мг фумаровой кислотыИК-спектр бисопролола фумарата Я  нтарная кислота/сукцинаты нтарная кислота/сукцинатыОписание призмы кристаллов (чаще моноклинной формы) или белый кристаллический порошок, без запаха Растворимость 1 г растворим в: 13 мл холодной воды; 1 мл горячей воды; Растворим в этаноле, этиловом эфире, ацетоне, метаноле; не растворим в толуоле, бензоле. Т плавл = 188оС pK1 = 4,207, pK2 = 5,635 при 25 ° C ФС по ГФ 14 М   етопролола сукцинат Этилметилгидроксипиридина сукцинат (собственно субстанция + 3 ЛФ) етопролола сукцинат Этилметилгидроксипиридина сукцинат (собственно субстанция + 3 ЛФ)ФС по ЕФ 8 издания Доксиламина сукцинат  В ЕФ 8 издания есть также сложные эфиры янтарной кислоты - такие как Метилпреднизолона гидрогенсукцинат или токоферола гидрогенсукцинат   Однако рассматривать последние две субстанции мы не будем ввиду того, что это не свободные поликарбоновые кислоты, а их эфиры, которые не являются темой курсовой работы В 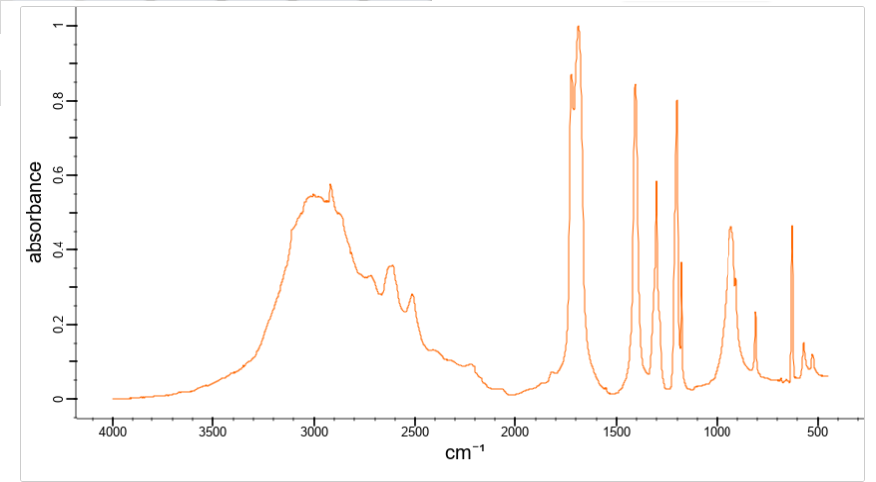 качестве метода подтверждения подлинности в случае метопролола сукцината и доксиламина гидрогенсукцината в ГФ 14 и ЕФ 8 издания соответственно указана только ИК-спектроскопия качестве метода подтверждения подлинности в случае метопролола сукцината и доксиламина гидрогенсукцината в ГФ 14 и ЕФ 8 издания соответственно указана только ИК-спектроскопияИ  К-спектр янтарной кислоты К-спектр янтарной кислотыИк-спектр метопролола сукцината (https://www.researchgate.net/figure/Metoprolol-Succinate-pure-drug-FTIR-spectrum_fig1_255909973 ) В случае этилметилгидроксипиридина сукцината – указана еще реакция с резорцином и серной кислотой (которая применима для всех его 3 ЛФ): В сухую пробирку вносят 5 мг субстанции и 10 мг резорцина, прибавляют 0.1 мл серной кислоты концентрированной и осторожно нагревают над пламенем горелки до тех пор, пока смесь не окрасится в темно-коричневый цвет. После охлаждения прибавляют 0,5 мл воды, затем натрия гидроксида раствор 10% до щелочной реакции, после чего доводят объем раствора водой до 20 мл; должно наблюдаться образование раствора оранжевого цвета, имеющего интенсивную зелёную флуоресценцию.  https://www.sielc.com/compound-succinic-acid.html Максимум поглощения в УФ-области = 208 нм (водный раствор) Винная кислота/Тартраты/Гидротартраты 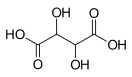 Непосредственно ФС о винной кислоте в ГФ 14 – нет, есть только ее соли, а вот в ЕФ 8 издания – есть, поэтому с данной ФС мы и ознакомимся Acidum tartaricum МНН: (2R, 3R) -2,3-Дигидроксибутандиовая кислота Содержание: от 99,5% до 101,0% (в пересчете на сухое вещество). Описание: белый или почти белый кристаллический порошок или бесцветные кристаллы. Растворимость: хорошо растворяется в воде, свободно растворяется в этаноле (96%). ИДЕНТИФИКАЦИЯ A. Раствор S (см. Тесты) является сильнокислым (2.2.4). Б. Он дает реакции тартратов. Р 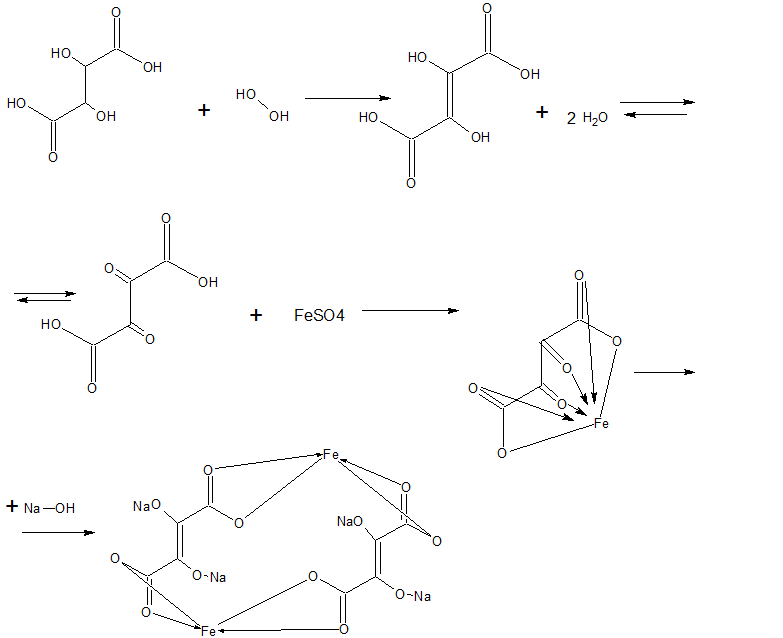 астворите около 15 мг исследуемого вещества в 5 мл воды или используйте 5 мл предписанного раствора. Добавьте 0,05 мл раствора сульфата двухвалентного железа с концентрацией 10 г / л и 0,05 мл разбавленного раствора перекиси водорода. Появляется временная желтая окраска. После исчезновения окраски по каплям добавляют разбавленный раствор гидроксида натрия. Получается фиолетовый или пурпурный цвет. астворите около 15 мг исследуемого вещества в 5 мл воды или используйте 5 мл предписанного раствора. Добавьте 0,05 мл раствора сульфата двухвалентного железа с концентрацией 10 г / л и 0,05 мл разбавленного раствора перекиси водорода. Появляется временная желтая окраска. После исчезновения окраски по каплям добавляют разбавленный раствор гидроксида натрия. Получается фиолетовый или пурпурный цвет.К 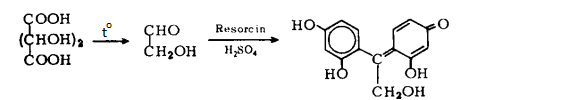 0,1 мл раствора исследуемого вещества, содержащего примерно 15 мг винной кислоты на миллилитр, или к 0,1 мл предписанного раствора добавить 0,1 мл раствора бромида калия с концентрацией 100 г / л, 0,1 мл 20 г / л раствора резорцина и 3 мл серной кислоты. Нагревают на водяной бане от 5 до 10 минут. Развивается темно-синий цвет. Дать остыть и вылить раствор в воду. Цвет изменится на красный. 0,1 мл раствора исследуемого вещества, содержащего примерно 15 мг винной кислоты на миллилитр, или к 0,1 мл предписанного раствора добавить 0,1 мл раствора бромида калия с концентрацией 100 г / л, 0,1 мл 20 г / л раствора резорцина и 3 мл серной кислоты. Нагревают на водяной бане от 5 до 10 минут. Развивается темно-синий цвет. Дать остыть и вылить раствор в воду. Цвет изменится на красный. К субстанции добавляют ацетат натрия и калия хлорид и аккуратно палочкой перемешивают раствор. Возникает белый осадок калия гидротартрата. ИСПЫТАНИЯ Раствор S. - растворите 5,0 г в дистиллированной воде R и доведите до 50 мл тем же растворителем. Внешний вид раствора. Раствор S прозрачный и не более интенсивно окрашен, чем контрольный раствор Y6. Удельное оптическое вращение: от + 12,0 до + 12,8 (высушенное вещество). Растворить 5,00 г в воде и разбавить тем же растворителем до 25,0 мл. Щавелевая кислота: максимум 350 частей на миллион в пересчете на безводную щавелевую кислоту. Растворить 0,80 г в 4 мл воды. Добавить 3 мл соляной кислоты и 1 г цинка в гранулах и кипятить 1 мин. Дать постоять 2 мин. Собирают жидкость в пробирку, содержащую 0,25 мл раствора фенилгидразина гидрохлорида концентрацией 10 г / л, и нагревают до кипения. Быстро охладить, перенести в мерный цилиндр, добавить равный объем соляной кислоты и 0,25 мл раствора феррицианида калия концентрацией 50 г / л. Встряхнуть и дать постоять 30 мин. Интенсивность розовой окраски исследуемого раствора не должна превышать интенсивность окраски стандартного раствора, приготовленного одновременно тем же способом с использованием 4 мл 0,1 г / л раствора щавелевой кислоты. Хлориды (2.4.4): максимум 100 ppm. Сульфаты (2.4.13): максимум 150 ppm. Кальций (2.4.3): максимум 200 ppm. Тяжелые металлы (2.4.8): максимум 10 частей на миллион. Потери при сушке (2.2.32): максимум 0,2%, определено на 1.000 г путем сушки в печи при 105 ° C. Сульфатная зола (2.4.14): не более 0,1%, определено на 1,0 г. 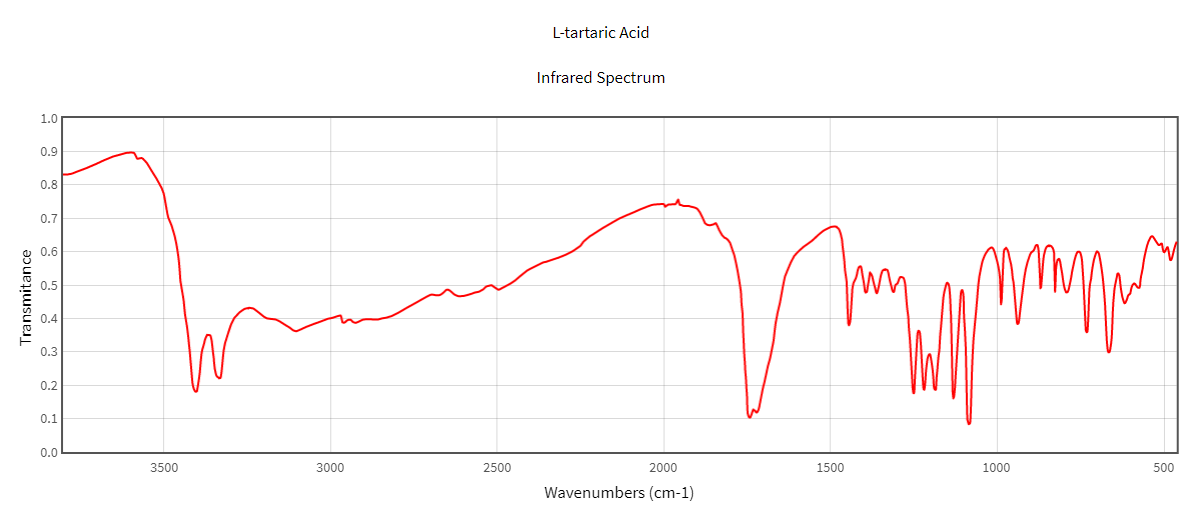 ИК-спектр винной кислоты Количественный анализ. Растворите 0,650 г в 25 мл воды. Титруйте 1 М гидроксидом натрия, используя 0,5 мл раствора фенолфталеина в качестве индикатора, до получения розового цвета. 1 мл 1 М гидроксида натрия эквивалентен 75,05 мг C4H6O6. С   убстанции, являющиеся солями винной кислоты убстанции, являющиеся солями винной кислотыметопролола гидротартрат норадреналина гидротартрат   П   латифиллина гидротартрат Дигидроэрготамина тартрат латифиллина гидротартрат Дигидроэрготамина тартратНикотина тартрат дигидрат Дигидрокодеина гидротартрат   Э  рготамина тартрат Калия гидротартрат рготамина тартрат Калия гидротартрат М  орантела гидротартрат Золпидема тартрат орантела гидротартрат Золпидема тартратРивастигмина гидротартрат Щавелевая кислота/оксалаты Именно ФС на щавелевую кислоту ни в ГФ 14, ни в ЕФ 8 – нет, поэтому мы рассмотрим общие сведения относительно данной дикарбоновой кислоты – физико-химические свойства и методы ее определения – в том числе и в субстанциях, являющихся ее солью. О  писание писаниеГигроскопические бесцветные кристаллы или белый порошок Растворимость Очень легко растворим в спирте, растворим в эфире, практически нерастворим в хлороформе, бензоле pKa1=1.46 pKa2=4.40 Всего (включая и ГФ 14, и ЕФ 8) есть 2 субстанции, содержащие щавелевую кислоту – это: Бриллиантовый зеленый (ГФ 14) Нафтидрофурила гидрогеноксалат (ЕФ 8)   Бриллиантовый зеленый Нафтидрофурила гидрогеноксалат Р 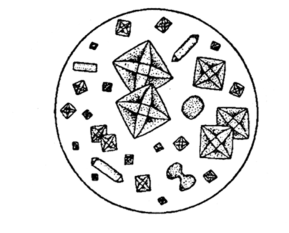 еакция подлинности именно на оксалат-ион указана только во второй субстанции – формирование осадка оксалата кальция, форму кристаллов которого (под микроскопом) вы можете видеть справа еакция подлинности именно на оксалат-ион указана только во второй субстанции – формирование осадка оксалата кальция, форму кристаллов которого (под микроскопом) вы можете видеть справаИз ФС на вторую субстанцию: Растворить 0,5 г в воде и разбавить тем же растворителем до 10 мл. Добавить 2,0 мл раствора хлорида кальция. Образуется белый осадок. Осадок растворяется после добавления 3,0 мл разведенной хлористоводородной кислоты. Чаще оксалат рассматривают как нежелательную примесь – как определяют долю щавелевой кислоты / её солей в этом случае рассмотрено в ФС «лимонная кислота» У 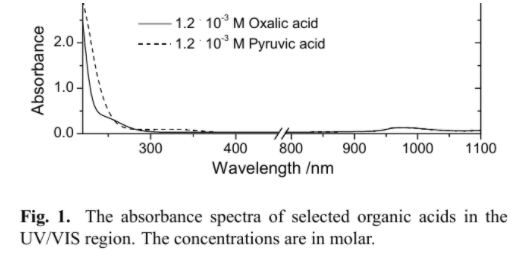 Ф-спектры щавелевой кислоты в воде и других растворителях Ф-спектры щавелевой кислоты в воде и других растворителях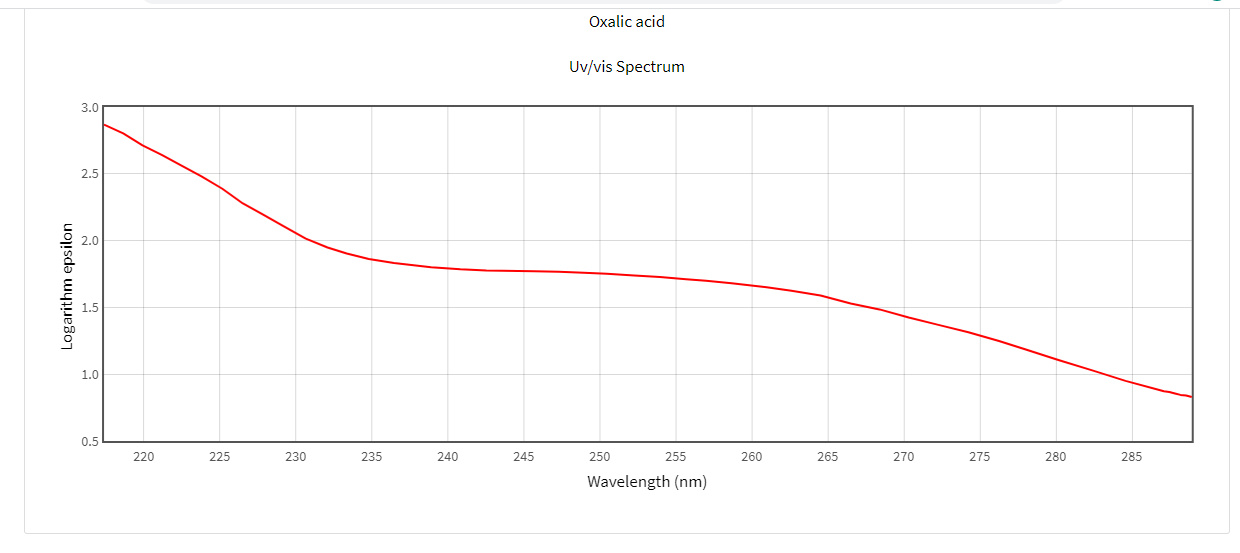 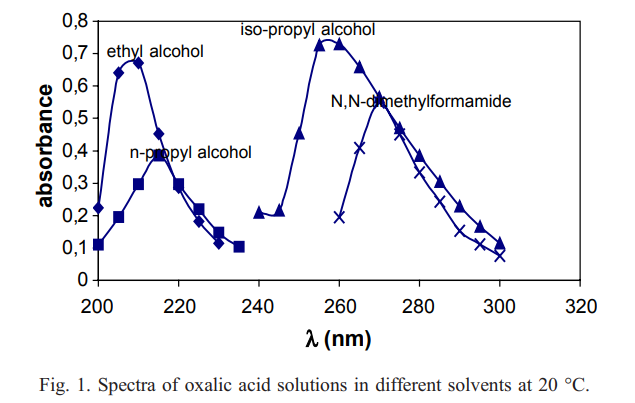 Малеиновая кислота/Малеаты (из ЕФ 8)  Малеиновая кислота содержит не менее 99,0% и не более 101,0% эквивалента (Z) -бутендиовой кислоты в пересчете на безводное вещество. Описание Белый или почти белый кристаллический порошок, Растворимость легко растворим в воде и спирте. Реакции Разбавьте 5 мл раствора S (см. Испытания) до 10 мл водой. pH разведения меньше 2. Изучите хроматограммы, полученные при испытании на фумаровую кислоту. Основное пятно на хроматограмме, полученной с контрольным раствором (b), по положению и размеру аналогично главному пятну на хроматограмме, полученной с контрольным раствором (a). Растворить 0,1 г в 10 мл воды (раствор а). К 0,3 мл раствора (а) добавляют раствор 10 мг резорцина в 3 мл серной кислоты. Нагревают на водяной бане в течение 15 мин; цвет не развивается. К 3 мл раствора (а) добавляют 1 мл бромной воды. Нагревают на водяной бане для удаления брома (15 мин), нагревают до кипения и охлаждают. К 0,2 мл этого раствора добавляют раствор 10 мг резорцина в 3 мл серной кислоты. Нагревают на водяной бане 15 мин. Развивается фиолетово-розовый цвет. Не указано в ЕФ – Обесцвечивание бромной воды Взаимодействие с карбонатом бария (2% раствором) с образованием белого осадка малеата бария, растворимого в разведенной хлористоводородной кислоте ИСПЫТАНИЯ Раствор S. Растворить 5,0 г в воде и разбавить тем же растворителем до 50 мл. Внешний вид раствора. Раствор S прозрачный и не более интенсивно окрашен, чем контрольный раствор Y7 Фумаровая кислота. Проверяют с помощью тонкослойной хроматографии , используя силикагель GF254 R в качестве вещества покрытия. Контрольный раствор (а). Растворите 0,5 г исследуемого вещества в ацетоне и доведите до 5 мл тем же растворителем. Контрольный раствор (б). Разбавьте 1 мл исследуемого раствора (а) до 50 мл ацетоном. Эталонный раствор (а). Растворите 20 мг CRS малеиновой кислоты в ацетоне и разбавьте до 10 мл тем же растворителем. Эталонный раствор (б). Растворите 15 мг CRS фумаровой кислоты в ацетоне и разбавьте до 10 мл тем же растворителем. Эталонный раствор (с). Смешайте 5 мл эталонного раствора (а) и 5 мл эталонного раствора (б). Нанесите отдельно на планшет 5 мкл тестовых растворов (а) и (б), 5 мкл эталонных растворов (а) и (б) и 10 мкл эталонного раствора (в). Проявите в ненасыщенном резервуаре на расстоянии 10 см, используя смесь из 12 объемов безводной муравьиной кислоты, 16 объемов хлороформа, 32 объемов бутанола и 44 объемов гептана. Сушат пластинку при 100 ° C в течение 15 мин и исследуют в УФ-свете при 254 нм. Любое пятно, соответствующее фумаровой кислоте, на хроматограмме исследуемого раствора (а) не более интенсивно, чем пятно на хроматограмме, полученной с эталонным раствором (b) (1,5%). Тест считается недействительным, если хроматограмма, полученная с эталонным раствором (c), не показывает два четко разделенных пятна. Ф   С по ГФ 14 (выделены) и ЕФ 8 С по ГФ 14 (выделены) и ЕФ 8Эналаприла малеат адреналина малеат М   епирамина малеат Эргометрина малеат епирамина малеат Эргометрина малеат  Флувоксамина малеат Тримипрамина малеат   Бромфенирамина малеат Левомепромазина малеат   Диметиндена малеат Тримебутина малеат   Прохлорперазина малеат Фенирамина малеат   Домперидона малеат дексхлорфенирамина малеат Сравнение определения подлинности различных поликарбоновых кислот Так или иначе, для всего ряда этих дикарбоновых кислот существует одна и та же реакция – с резорцином и серной кислотой при нагревании Окраски растворов после взаимодействия с резорцином и серной кислотой
В случае с непредельными кислотами – отличить от насыщенных их можно по реакции с бромной водой, которую они и обесцвечивают Также, с практической точки зрения имеет немаловажное значение и разница в растворимостях конкретных солей конкретных субстанций: так, например, одна из выделяющихся разниц между метопролола тартратом и метопролола сукцинатом – растворимость в спирте: сукцинат плохо растворяется в нем, а тартрат – легко. В том случае, если субстанция содержит несколько поликарбоновых кислот, то первостепенное значение имеет возможность их одновременного определения с высокой эффективностью – такую возможность могут предоставить методы хроматографии (ТСХ/ВЭЖХ) и капиллярного электрофореза Одновременное определение поликарбоновых кислот Н 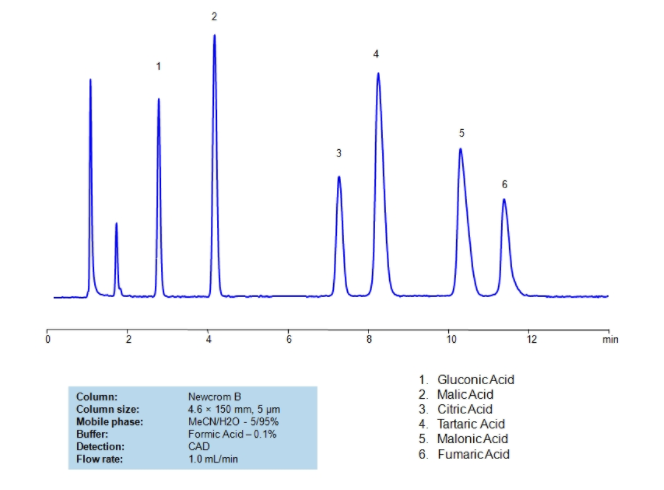 а примере ВЭЖХ а примере ВЭЖХН 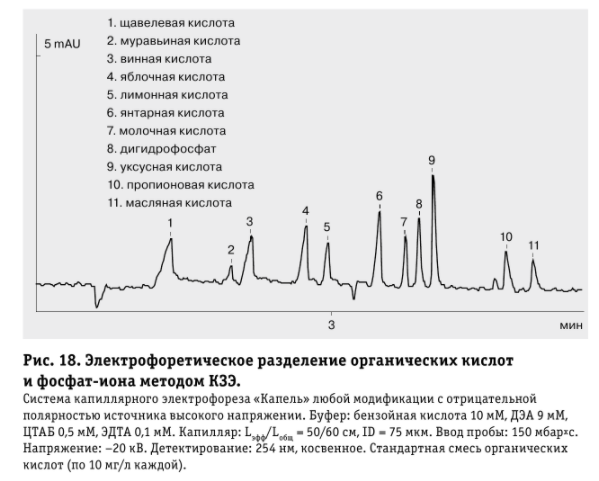 а примере капиллярного электрофореза а примере капиллярного электрофорезаВозможные методы анализа, которые не указаны в ГФ ВЭЖХ-МС ГХ-МС ЯМР-спектроскопия Капиллярный электрофорез Хотелось бы отметить, что одним из немаловажных преимуществ является свободный доступ к библиотекам по МС и ЯМР-спектроскопии, в которых указано, например, какие показатели M/Z должны быть для конкретной субстанции Капиллярный электрофорез и его модификация – мицеллярная электрокинетическая хроматография – могут стать прекрасными методами определения подлинности, однако надлежащие методики для этого еще не валидированы и в целом – данные методы менее распространены, поэтому на данный момент эти методики не пользуются особой популярностью. Ик-спектроскопия Общие особенности  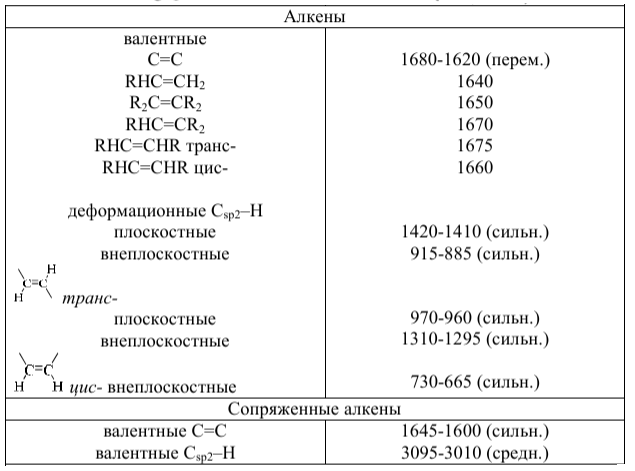 ИК-спектроскопия является достаточно удобным и относительно простым методом, который позволяет одновременно с высокой вероятностью удостовериться в надлежащей подлинности и отсутствии недопустимых примесей в субстанции Так, например, мы можем сразу наглядно видеть на этих двух спектрах, какой из двух кислот (фумаровой или щавелевой) какой спектр принадлежит – на первом спектр выражен пик транс-плоскостных и –внеплоскостных колебаний, а на втором – еще более, чем первом – выражен пик валентных С=О колебаний (что связано с наличие двух оксо групп, а не одной) 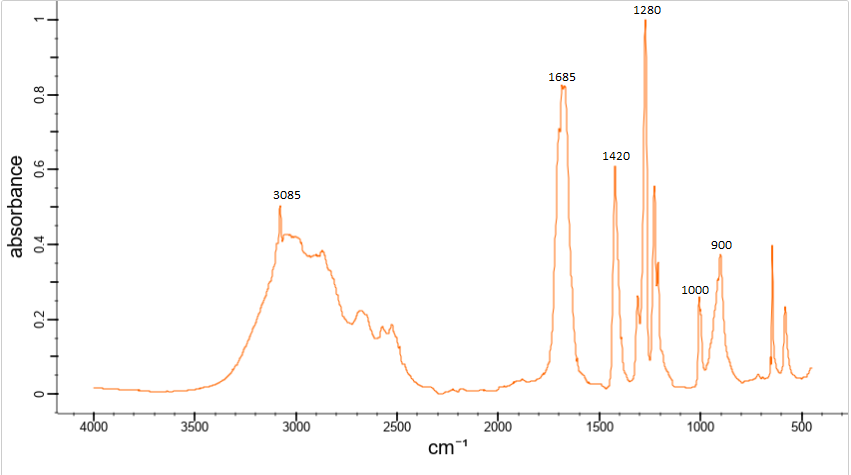 ИК-спектр соли фумаровой кислоты 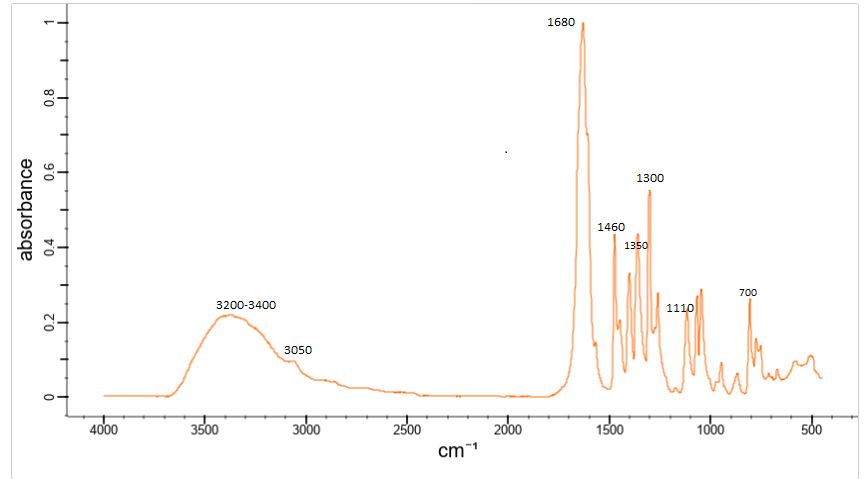 ИК-спектр щавелевой кислоты Выводы Список литературы |