Ответы на билеты биохимия 2012. Белок это последовательность ак, связанных друг с другом пептидными связями
 Скачать 5.15 Mb. Скачать 5.15 Mb.
|

|
1. Метаболизм феиилаланина Основное количество фенилаланина расходуется по 2 путям: 1)включается в белки;2)превращается в тирозин. Превращение фенилаланина в тирозин прежде всего необходимо для удаления избытка фенилаланина, так как высокие концентрации его токсичны для клеток. Образование тирозина не имеет большого значения, так как недостатка этой аминокислоты в клетках практически не бывает. Основной путь метаболизма фенилаланина начинается с его гидроксилирования (рис. 9-29), в результате чего образуется тирозин. Эта реакция катализируется специфической монооксиге-назой - фенилаланингидр(жсилазой, кофермен-том которой служит тетрагидробиоптерин (Н4БП). Активность фермента зависит также от наличия Fe2+. Реакция необратима. Н4БП в результате реакции окисляется в дигидробиоптерин (Н2БП). Регенерация последнего происходит при участии дигидроптеридинредуктазы с использованием NADPH + H+. 2. Особенности обмена тирозина в разных тканях. Обмен тирозина значительно сложнее, чем обмен фенилаланина. Кроме использования в синтезе белков, тирозин в разных тканях выступает предшественником таких соединений, как катехоламины, тироксин, меланины, и ка-таболизируется до СО2 и Н2О. Катаболизм тирозина в печени. В печени происходит катаболизм тирозина до конечных продуктов. Специфический путь катаболизма включает несколько ферментативных реакций, завершающихся образованием фумарата и ацетоацетата. Т 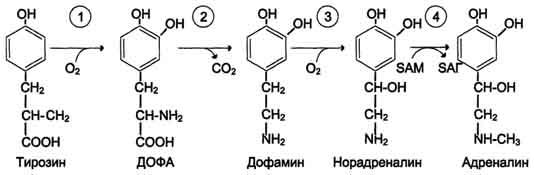 рансаминирование тирозина с ос-кетоглутаратом катализирует тирозинаминотрансфе-раза (кофермент ПФ) - индуцируемый фермент печени млекопитающих. В результате образуется п-гидроксифенилпируват. В реакции окисления п-гидроксифенилпирувата в гомогентизиновую кислоту происходит декарбоксилирование, гидроксилирование ароматического кольца и миграция боковой цепи. Реакцию катализирует фермент п-гидроксифенилпируватдиоксигеназа, кофакторами которого выступают витамин С и Fe2+. Превращение гомогентизиновой кислоты в фумарилацетоацетат сопровождается расщеплением ароматического кольца. Эта реакция катализируется диоксигеназой гомогентизиновой кислоты, в качестве кофермента содержащей Fe2+. Обмен фенилаланина и тирозина связан со значительным количеством реакций гидроксилирования, которые катализируют оксигеназы. Ферменты оксигеназы (гидроксилазы) используют молекулу О2 и кофермент-донор водорода (чаще - Н4БП). Для катализа оксигеназам не обходимы кофакторы - Fe2+ или гем (для некоторых - Сu+), а для многих ещё и витамин С. Оксигеназы делят на 2 группы: Монооксигеназы - один атом О2 присоединяют к продукту реакции, другой используют для образования Н2О; Диоксигеназы - оба атома О2 используют для образования продукта реакции. Почти все процессы расщепления ароматических колец в биологических системах катализируются диоксигеназами. В результате разрыва бензольного кольца образуется малеилацетоацетат, который в процессе цис- и транс-изомеризации превращается в фумарилацетоацетат. рансаминирование тирозина с ос-кетоглутаратом катализирует тирозинаминотрансфе-раза (кофермент ПФ) - индуцируемый фермент печени млекопитающих. В результате образуется п-гидроксифенилпируват. В реакции окисления п-гидроксифенилпирувата в гомогентизиновую кислоту происходит декарбоксилирование, гидроксилирование ароматического кольца и миграция боковой цепи. Реакцию катализирует фермент п-гидроксифенилпируватдиоксигеназа, кофакторами которого выступают витамин С и Fe2+. Превращение гомогентизиновой кислоты в фумарилацетоацетат сопровождается расщеплением ароматического кольца. Эта реакция катализируется диоксигеназой гомогентизиновой кислоты, в качестве кофермента содержащей Fe2+. Обмен фенилаланина и тирозина связан со значительным количеством реакций гидроксилирования, которые катализируют оксигеназы. Ферменты оксигеназы (гидроксилазы) используют молекулу О2 и кофермент-донор водорода (чаще - Н4БП). Для катализа оксигеназам не обходимы кофакторы - Fe2+ или гем (для некоторых - Сu+), а для многих ещё и витамин С. Оксигеназы делят на 2 группы: Монооксигеназы - один атом О2 присоединяют к продукту реакции, другой используют для образования Н2О; Диоксигеназы - оба атома О2 используют для образования продукта реакции. Почти все процессы расщепления ароматических колец в биологических системах катализируются диоксигеназами. В результате разрыва бензольного кольца образуется малеилацетоацетат, который в процессе цис- и транс-изомеризации превращается в фумарилацетоацетат. Гидролиз фумарилацетоацетата при действии фумарилацетоацетатгидролазы приводит к образованию фумарата и ацетоацетата. Фумарат может окисляться до СО2 и Н2О или использоваться для глюконеогенеза. Ацетоацетат - кетоновое тело, окисляемое до конечных продуктов с выделением энергии. Превращение тирозина в меланоцитах. В пигментных клетках (меланоцитах) тирозин выступает предшественником тёмных пигментов - меланинов. Среди них преобладают 2 типа: эумеланины и феомеланины. Эумеланины (чёрного и коричневого цвета) - нерастворимые высокомолекулярные гетерополимеры 5,6-дигидроксииндола и некоторых его предшественников. Феомеланины - жёлтые или красновато-коричневые полимеры, растворимые в разбавленных щелочах. Находятся они, в основном, в составе волос. Меланины присутствуют в сетчатке глаз. Цвет кожи зависит от распределения меланоцитов и количества в них разных типов меланинов. Первую реакцию - превращение тирозина в ДОФА - катализирует тирозиназа, использующая в качестве кофактора ионы Сu+. Превращение тирозина в щитовидной железе. В щитовидной железе синтезируются и выделяются гормоны йодтиронины: тироксин (тет-райодтиронин) и трийодтиронин. Эти гормоны представляют собой йодированные остатки тирозина, которые попадают в клетки щитовидной железы через базальную мембрану. Превращения тирозина в надпочечниках и нервной ткани (синтез катехоламинов) .В мозговом веществе надпочечников и нервной ткани тирозин является предшественником катехоламинов (дофамина, норадреналина и адреналина). При образовании катехоламинов, которое происходит в нервной ткани и надпочечниках, и меланина в меланоцитах промежуточным продуктом служит диоксифенилаланин (ДОФА) . Однако гидроксилирование тирозина в клетках различных типов катализируется различными ферментами: Тирозиназа в меланоцитах является Сu+-зависимым ферментом (см. выше). Тирозингидроксилаза (1) в надпочечниках и ка-техоламинергических нейронах не нуждается в ионах меди. Это - Fе2+-зависимый фермент, аналогично фенилаланингидроксилазе в качестве кофермента использующий Н4БП. Физиологическая роль тирозингидроксилазы чрезвычайно велика, так как этот фермент является регуляторным и определяет скорость синтеза катехоламинов. Активность тирозингидроксилазы значительно изменяется в результате: 1. Аллостерической регуляции (ингибитор - норадреналин); 2.Фосфорилирования/дефосфорилирования: в результате фосфорилирования с участием протеинкиназы А снижаются Кm для кофермента Н4БП и сродство фермента к норадреналину, в результате чего происходит активация тирозингидроксилазы. К  оличество фермента регулируется на уровне транскрипции. ДОФА-декарбоксилаза (2) (кофермент - ПФ) катализирует образование дофамина, который при участии дофамингидроксилазы (3) (монооксигеназы) превращается в норадреналин. Для функционирования фермента необходимы ионы Сu+, витамин С и тетрагидробиоптерин. В мозговом веществе надпочечников фенилэтаноламин-N-метилтрансфераза (4) катализирует метилирование норадреналина, в результате чего образуется адреналин. Источником метальной группы служит &АМ. Дофамин и норадреналин служат медиаторами в синаптической передаче нервных импульсов, а адреналин - гормон широкого спектра действия, регулирующий энергетический обмен. Одна из функций катехоламинов - регуляция деятельности ССС.Известно несколько наследственных заболеваний, связанных с дефектом ферментов обмена фенилаланина и тирозина в разных тканях. Фенилкетонурия. В печени здоровых людей небольшая часть фенилаланина (∼10%) превращается в фенил-лактат и фенилацетилглутамин. Этот путь катаболизма фенилаланина становится главным при нарушении основного пути - превращения в тирозин, катализируемого фенил-аланингидроксилазой. Такое нарушение сопровождается гиперфенилаланинемией и повышением в крови и моче содержания метаболитов альтернативного пути: фенилпирувата, фенилацетата, фениллактата и фенилацетилглу-тамина. Дефект фенилаланингидроксилазы приводит к заболеванию фенилкетонурия (ФКУ). Выделяют 2 формы ФКУ: Классическая ФКУ - наследственное заболевание, связанное с мутациями в гене фенилаланингидроксилазы, которые приводят к снижению активности фермента или полной его инактивации. При этом концентрация фенилаланина повышается в крови в 20-30 раз (в норме - 1,0-2,0 мг/дл), в моче - в 100-300 раз по сравнению с нормой (30 мг/дл). Концентрация фенилпирувата и фениллактата в моче достигает 300-600 мг/дл при полном отсутствии в норме. Наиболее тяжёлые проявления ФКУ - нарушение умственного и физического развития, судорожный синдром, нарушение пигментации. При отсутствии лечения больные не доживают до 30 лет. Частота заболевания - 1:10 000 новорождённых. Заболевание наследуется по аутосомно-рецессивному типу. Тяжёлые проявления ФКУ связаны с токсическим действием на клетки мозга высоких концентраций фенилаланина, фенилпирувата, фениллактата. Большие конце нтрации фенилаланина ограничивают транспорт тирозина и триптофана через гематоэнцефаличеекий барьер и тормозят синтез нейро-медиаторов (дофамина, норадреналина, серотонина). оличество фермента регулируется на уровне транскрипции. ДОФА-декарбоксилаза (2) (кофермент - ПФ) катализирует образование дофамина, который при участии дофамингидроксилазы (3) (монооксигеназы) превращается в норадреналин. Для функционирования фермента необходимы ионы Сu+, витамин С и тетрагидробиоптерин. В мозговом веществе надпочечников фенилэтаноламин-N-метилтрансфераза (4) катализирует метилирование норадреналина, в результате чего образуется адреналин. Источником метальной группы служит &АМ. Дофамин и норадреналин служат медиаторами в синаптической передаче нервных импульсов, а адреналин - гормон широкого спектра действия, регулирующий энергетический обмен. Одна из функций катехоламинов - регуляция деятельности ССС.Известно несколько наследственных заболеваний, связанных с дефектом ферментов обмена фенилаланина и тирозина в разных тканях. Фенилкетонурия. В печени здоровых людей небольшая часть фенилаланина (∼10%) превращается в фенил-лактат и фенилацетилглутамин. Этот путь катаболизма фенилаланина становится главным при нарушении основного пути - превращения в тирозин, катализируемого фенил-аланингидроксилазой. Такое нарушение сопровождается гиперфенилаланинемией и повышением в крови и моче содержания метаболитов альтернативного пути: фенилпирувата, фенилацетата, фениллактата и фенилацетилглу-тамина. Дефект фенилаланингидроксилазы приводит к заболеванию фенилкетонурия (ФКУ). Выделяют 2 формы ФКУ: Классическая ФКУ - наследственное заболевание, связанное с мутациями в гене фенилаланингидроксилазы, которые приводят к снижению активности фермента или полной его инактивации. При этом концентрация фенилаланина повышается в крови в 20-30 раз (в норме - 1,0-2,0 мг/дл), в моче - в 100-300 раз по сравнению с нормой (30 мг/дл). Концентрация фенилпирувата и фениллактата в моче достигает 300-600 мг/дл при полном отсутствии в норме. Наиболее тяжёлые проявления ФКУ - нарушение умственного и физического развития, судорожный синдром, нарушение пигментации. При отсутствии лечения больные не доживают до 30 лет. Частота заболевания - 1:10 000 новорождённых. Заболевание наследуется по аутосомно-рецессивному типу. Тяжёлые проявления ФКУ связаны с токсическим действием на клетки мозга высоких концентраций фенилаланина, фенилпирувата, фениллактата. Большие конце нтрации фенилаланина ограничивают транспорт тирозина и триптофана через гематоэнцефаличеекий барьер и тормозят синтез нейро-медиаторов (дофамина, норадреналина, серотонина). Вариантная ФКУ (коферментзависимая гиперфенилаланинемия) - следствие мутаций в генах, контролирующих метаболизм Н4БП. Клинические проявления - близкие, но не точно совпадающие с проявлениями классической ФКУ. Частота заболевания - 1-2 случая на 1 млн новорождённых. Н4БП необходим для реакций гидроксилирования не только фенилаланина, но также тирозина и триптофана, поэтому при недостатке этого кофермента нарушается метаболизм всех 3 аминокислот, в том числе и синтез ней-ромедиаторов. Заболевание характеризуется тяжёлыми неврологическими нарушениями и ранней смертью ("злокачественная" ФКУ). Прогрессирующее нарушение умственного и физического развития у детей, больных ФКУ, можно предотвратить диетой с очень низким содержанием или полным исключением фенилаланина. Если такое лечение начато сразу после рождения ребёнка, то повреждение мозга предотвращается. Считается, что ограничения в питании могут быть ослаблены после 10-летнего возраста (окончание процессов миелинизации мозга). Некоторые нарушения катаболизма тирозина в печени приводят к тирозинемии и тирозинурии: Тирозинемия типа 1 (тирозиноз). Причиной заболевания является, вероятно, дефект фермента фумарилацетоацетатгидролазы, катализирующего расщепление фумарилацетоа-цетата на фумарат и ацетоацетат (рис. 9-28). Накапливающиеся метаболиты снижают активность некоторых ферментов и транспортных систем аминокислот. Патофизиология этого нарушения достаточно сложна. Острая форма тирозиноза характерна для новорождённых. Клинические проявления - диарея, рвота, задержка в развитии. Без лечения дети погибают в возрасте 6-8 мес из-за развивающейся недостаточности печени. Хроническая форма характеризуется сходными, но менее выраженными симптомами. Гибель наступает в возрасте 10 лет. Содержание тирозина в крови у больных в несколько раз превышает норму. Для лечения используют диету с пониженным содержанием тирозина и фенилаланина. Тирозинемия типа II (синдром Рихнера-Ханхорта). Причина - дефект фермента тирозина-минотрансферазы. Концентрация тирозина в крови больных повышена. Для заболевания характерны поражения глаз и кожи, умеренная умственная отсталость, нарушение координации движений. Тирозинемия новорождённых (кратковременная). Заболевание возникает в результате снижения активности фермента п-гидроксифенилпируватдиоксигеназы, превращающего п-гидроксифенилпируват в гомогентизиновую кислоту. В результате в крови больных повышается концентрация п-гидроксифенилацетата, тирозина и фенил-аланина. При лечении назначают бедную белком диету и витамин С. Алкаптонурия ("чёрная моча"). Причина заболевания - дефект диоксигеназы гомогентизиновой кислоты. Для этой болезни характерно выделение с мочой большого количества гомогентизиновой кислоты, которая, окисляясь кислородом воздуха, образует тёмные пигменты алкаптоны. Клиническими проявлениями болезни, кроме потемнения мочи на воздухе, являются пигментация соединительной ткани (охроноз) и артрит. Частота - 2-5 случаев на 1 млн новорождённых. Альбинизм. Причина метаболического нарушения - врождённый дефект тирозиназы. Этот фермент катализирует превращение тирозина в ДОФА в меланоцитах. В результате дефекта тирозиназы нарушается синтез пигментов меланинов. Клиническое проявление альбинизма (от лат. albus - белый) - отсутствие пигментации кожи и волос. У больных часто снижена острота зрения, возникает светобоязнь. Длительное пребывание таких больных под открытым солнцем приводит к раку кожи. Частота заболевания 1:20 000. Нарушение синтеза катехоламинов может вызывать различные нервно-психические заболевания, причём патологические отклонения наблюдаются как при снижении, так и при увеличении их количества. Болезнь Паркинсона. Заболевание развивается при недостаточности дофамина в чёрной субстанции мозга. Это одно из самых распространённых неврологических заболеваний (частота 1:200 среди людей старше 60 лет). При этой патологии снижена активность тирозингидроксилазы, ДОФА-декарбоксилазы. Заболевание сопровождается тремя основными симптомами: акинезия (скованность движений), ригидность (напряжение мышц), тремор (непроизвольное дрожание). Дофамин не проникает через гематоэнцефалический барьер и как лекарственный препарат не используется. Для лечения паркинсонизма предлагаются следующие принципы: *заместительная терапия препаратами-предшественниками дофамина (производными ДОФА) - леводопа, мадопар, наком и др. *подавление инактивации дофамина ингибиторами МАО (депренил, ниаламид, пиразидол и др.). Депрессивные состояния часто связаны со снижением в нервных клетках содержания дофамина и норадреналина. Гиперсекреция дофамина в височной доле мозга наблюдается при шизофрении. Значение глицина: «+» образование гемма, переходит в серин, образует креатин, желчные кислоты, глутатион; «-» превращается в щевелеуксусную кислоту-> оксалаты(соли в почках => камни)Значение серина: из него синтезируется пируват, цистеин, сфинголипиды, фосфолипиды, 3-фосфоглоцерат-> глюкоза Значение метионина- необходим для синтеза белков, участвует в реакции дезаминирования, является источником атома серы для синтеза цистеина. Роль реакции превращения серина в глицин состоит в образовании активной формы тетрагидрофолиевой кислоты – N5,N10-метилен-ТГФК. Одновременно данная реакция является первой на пути катаболизма серина. Несмотря на простоту строения, глицин и серин являются весьма востребованными аминокислотами в клетках. Благодаря взаимопревращению перечень возможных путей метаболизма этих аминокислот еще больше расширяется. О  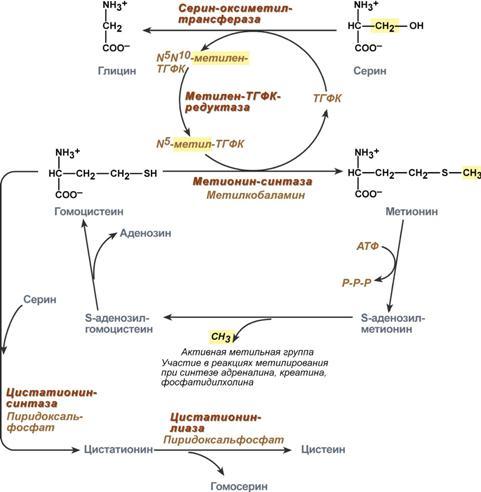  бразованный в реакции распада серина до глицина N5,N10-метилен-ТГФК при участии фермента метилен-ТГФК-редуктазы превращается в N5-метил-ТГФК. Ее метильный остаток участвует в метионин-синтазной реакции реметилирования гомоцистеина в метионин. В печени, кроме метил-ТГФК, источником метильной группы может быть вещество бетаин (триметилглицин).Метионин впоследствии присоединяет аденозильный остаток и превращается в активную форму метионина – S-аденозилметионин, участвующий во многих реакциях метилирования, в частности, при синтезе креатина, карнитина, фосфатидилхолина, адреналина. В результате перемещения метильной группы и отщепления аденозина остается гомоцистеин, имеющий два пути метаболизма:Первый путь превращений гомоцистеина – реметилирование до метионина и вновь участие в реакциях метилирования и синтезе веществ. Второй путь – взаимодействие с серином при участии цистатионин-синтазы, превращение в цистатионин с последующим распадом в цистеин и гомосерин. бразованный в реакции распада серина до глицина N5,N10-метилен-ТГФК при участии фермента метилен-ТГФК-редуктазы превращается в N5-метил-ТГФК. Ее метильный остаток участвует в метионин-синтазной реакции реметилирования гомоцистеина в метионин. В печени, кроме метил-ТГФК, источником метильной группы может быть вещество бетаин (триметилглицин).Метионин впоследствии присоединяет аденозильный остаток и превращается в активную форму метионина – S-аденозилметионин, участвующий во многих реакциях метилирования, в частности, при синтезе креатина, карнитина, фосфатидилхолина, адреналина. В результате перемещения метильной группы и отщепления аденозина остается гомоцистеин, имеющий два пути метаболизма:Первый путь превращений гомоцистеина – реметилирование до метионина и вновь участие в реакциях метилирования и синтезе веществ. Второй путь – взаимодействие с серином при участии цистатионин-синтазы, превращение в цистатионин с последующим распадом в цистеин и гомосерин.3 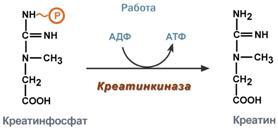 4. Синтез креатина: последовательность реакций, значение креатинфосфата. Физиологическая креатинурия. Значение креатинкиназы и креатинина в диагностике. 4. Синтез креатина: последовательность реакций, значение креатинфосфата. Физиологическая креатинурия. Значение креатинкиназы и креатинина в диагностике.Креатин – вещество скелетных мышц, миокарда, нервной ткани. В виде креатинфосфата креатин является "депо" макроэргических связей, используется для быстрого ресинтеза АТФ во время работы клетки. О    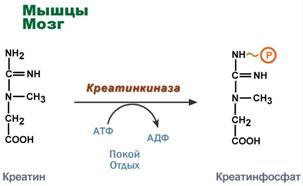 собенно показательна роль креатина в мышечной ткани. Креатинфосфат обеспечивает ресинтез АТФ в первые секунды работы (5 10 сек), когда ни анаэробный гликолиз, ни аэробное окисление глюкозы и жирных кислот еще не активировано, и кровоснабжение мышцы не увеличено. В клетках нервной ткани креатинфосфат поддерживает жизнеспособность клеток при отсутствии кислорода. При мышечной работе ионы Са2+, высвободившиеся из саркоплазматического ретикулума, являются активаторами креатинкиназы. Реакция еще интересна тем, что на ее примере можно наблюдать обратную положительную связь — активацию фермента продуктом реакции креатином. Это позволяет избежать снижения скорости реакции по ходу работы, которое должно было бы произойти по закону действующих масс из-за снижения концентрации креатинфосфата в работающих мышцах. Около 3% креатинфосфата постоянно в реакции неферментативного дефосфорилирования превращается в креатинин. Количество креатинина, выделяемое здоровым человеком в сутки, всегда почти одинаково и зависит только от объема мышечной массы. Синтез креатина идет последовательно в почках и печени в двух трансферазных реакциях. По окончании синтеза креатин с током крови доставляется в мышцы или мозг. Здесь при наличии энергии АТФ (во время покоя или отдыха) он фосфорилируется с образованием креатинфосфата,который после дефосфорилирования (необратимая реакция) превращается в креатинин, выделяющийся с мочой. собенно показательна роль креатина в мышечной ткани. Креатинфосфат обеспечивает ресинтез АТФ в первые секунды работы (5 10 сек), когда ни анаэробный гликолиз, ни аэробное окисление глюкозы и жирных кислот еще не активировано, и кровоснабжение мышцы не увеличено. В клетках нервной ткани креатинфосфат поддерживает жизнеспособность клеток при отсутствии кислорода. При мышечной работе ионы Са2+, высвободившиеся из саркоплазматического ретикулума, являются активаторами креатинкиназы. Реакция еще интересна тем, что на ее примере можно наблюдать обратную положительную связь — активацию фермента продуктом реакции креатином. Это позволяет избежать снижения скорости реакции по ходу работы, которое должно было бы произойти по закону действующих масс из-за снижения концентрации креатинфосфата в работающих мышцах. Около 3% креатинфосфата постоянно в реакции неферментативного дефосфорилирования превращается в креатинин. Количество креатинина, выделяемое здоровым человеком в сутки, всегда почти одинаково и зависит только от объема мышечной массы. Синтез креатина идет последовательно в почках и печени в двух трансферазных реакциях. По окончании синтеза креатин с током крови доставляется в мышцы или мозг. Здесь при наличии энергии АТФ (во время покоя или отдыха) он фосфорилируется с образованием креатинфосфата,который после дефосфорилирования (необратимая реакция) превращается в креатинин, выделяющийся с мочой.Е  сли синтез креатина опережает возможности его фиксации в мышечной ткани, то развивается креатинурия – появление креатина в моче. Физиологическая креатинурия наблюдается в первые годы жизни ребенка. Иногда к физиологической относят и креатинурию стариков, которая возникает как следствие атрофии мышц и неполного использования образующегося в печени креатина. При заболеваниях мышечной системы (при миопатии или прогрессирующей мышечной дистрофии) в моче наблюдаются наибольшие концентрации креатина – патологическая креатинурия. сли синтез креатина опережает возможности его фиксации в мышечной ткани, то развивается креатинурия – появление креатина в моче. Физиологическая креатинурия наблюдается в первые годы жизни ребенка. Иногда к физиологической относят и креатинурию стариков, которая возникает как следствие атрофии мышц и неполного использования образующегося в печени креатина. При заболеваниях мышечной системы (при миопатии или прогрессирующей мышечной дистрофии) в моче наблюдаются наибольшие концентрации креатина – патологическая креатинурия.Креатинин является конечным продуктом азотистого обмена. Образуется в мышечной ткани из креатинфосфата. Креатинин попадает в мочу преимущественно путем клубочковой фильтрации и в крайне небольшом количестве за счет активной канальцевой секреции. Норма: мужчины 7,1-17,7 ммоль/сут, женщины 5,3 15,9 Клиникодиагностическое значение. Выводимое количество мало зависит от содержания белков в диете, а связано с объемом мышечной ткани и ее активностью. Увеличение концентрации креатинина может быть связано с повышенной физической активностью, с лихорадочными состояниями, отмечается при выраженной недостаточности функции печени, при сахарном диабете, инфекциях. Снижение обнаруживается при голодании, у больных с мышечной атрофией, с дегенерацией и амилоидозом почек, лейкемией. |