Семинар 2. Фармацевтическая разработка (ФР)
 Скачать 0.54 Mb. Скачать 0.54 Mb.
|

 Фармацевтическая разработка (ФР) - это комплексное исследование компонентного состава (лекарственных субстанций и вспомогательных веществ), изучение их спецификаций, выбора лекарственной формы, а также оптимизации технологического процесса, упаковки, обоснование показателей качества и спецификации готового лекарственного средства (ГЛС). Фармацевтическая разработка (ФР) - это комплексное исследование компонентного состава (лекарственных субстанций и вспомогательных веществ), изучение их спецификаций, выбора лекарственной формы, а также оптимизации технологического процесса, упаковки, обоснование показателей качества и спецификации готового лекарственного средства (ГЛС).ФР целеустремленно определяет наиболее важные стадии, которые контролируются при рутинном производстве, и формирует качество ГЛС. ФР гарантирует высокую вероятность того, что каждая единица каждой серии препарата промышленного производства будет иметь качество, которое будет отвечать предвиденному назначению с той эффективностью, безопасностью, качеством, которое установлено при клинических исследованиях ГЛС. Идеи и положения ФР максимально надежно реализуются путем внедрения надлежащих практик (GMP, GLP,GCP,GDP). ФР есть обязательной составной частью регистрационного досье на ГЛС. Основными объектами исследований ФР есть компоненты ГЛС, лекарственная форма, технологический процесс, упаковочные материалы и валидация процесса производства. К компонентам ГЛС относятся лекарственные и вспомогательные вещества, количественные показатели качества которых устанавливаются экспериментально. Лекарственное вещество (субстанция) при производстве препарата рассматривается как терапевтически активный ингредиент, выбор которого осуществляется с учетом стабильности, биологической активности и наличия примесей. Важным есть исследования совместимости субстанции со вспомогательными веществами в лекарственной форме как на стадии производства, так и на протяжении срока хранения. В зависимости от состава лекарственной формы между субстанцией и вспомогательными веществами могут происходить взаимодействия, при которых возможно получение соединений-включений, комплексов и т.п.. Комплексное изучение физико-химических характеристик лекарственного вещества на этапе ФР может использоваться при обосновании выбора метода оценки эффективности и безопасности ГЛС. ФР дает научное обоснование выбора каждого вспомогательного вещества, которые разрешены для применения в производстве ГЛС. Вспомогательные вещества образовывают единую с лекарственным веществом систему, свойства которой определяются и направляются на обеспечение необходимой терапевтической эффективности препарата. Выбор вспомогательных веществ при ФР проводится с учетом отсутствия токсичного действия, взаимодействия с лекарственным веществом, уровня стабильности, технологичности, отсутствия взаимодействия с материалами первичной упаковки и технологическим оборудованием, влияния на органолептические свойства препарата и соответствия по показателям химической и микробиологической чистоты. Выбор вспомогательных веществ, их количественные и качественные характеристики должны отвечать предвиденной цели, технологическому процессу, условиям и сроку хранения ГЛС. На основании экспериментальных данных ФР определяет вид и состав лекарственной формы, которая обеспечивает оптимальный терапевтический эффект лекарственного вещества при минимуме побочного действия, фармакологическую рациональность, а также удобство при хранении и использовании ГЛС. В процессе ФР изучаются микробиологические характеристики, категории микробиологической чистоты , вспомогательных веществ, обосновываются посерийные или периодические испытания микробиологической чистоты ГЛС. Показатель качества «Микробиологическая чистота» вводится к спецификации на ГЛС. ФР исследует критические параметры и критерии приемлемости технологического процесса производства. На начальных стадиях разработки проводится анализ экспериментальных данных, которые получены на лабораторных сериях препарата. На этапе обрабатывания технологического процесса используются опытно-промышленные серии препарата, объем которых мажет составлять наименьше 10% от объема промышленной серии. Результаты исследований приводятся в регистрационном досье на ГЛС. Разработка новых препаратов включает в себя ряд последовательных этапов. Первый этап направлен на поиск перспективных соединений, возможно, обла дающих лечебным действием. Второй этап – это доклиническое изучение биологической ак тивности обозначенных к дальнейшему исследованию веществ. Доклиническое изучение вещества разделяется на фармакологическое и токсикологическое. Цель фармакологических исследований – определение не только терапевтической эффективности препарата и его влияния на системы организма, но и возможных побочных реакций, связанных с фармакологической активностью. При токсикологических исследованиях устанавливают характер и возможные повреждающие воздействия на организм экспериментальных животных. Выделяют три этапа токсикологических исследований: 1) изучение токсичности препарата при однократном введении; 2) определение хронической токсичности вещества при повторном введении на протяжении 1 года и больше; 3) установление специфического влияния соединения (онкогенность, мутагенность, воздействие на плод и др.). Третий этап – клинические испытания нового лекарственного вещества. Проводится оценку терапевтической или профилактической эффективности, переносимости, установление доз и схем применения препарата, а также сравнительных характеристик с другими лекарственными средствами. В процессе клинических испытаний выделяют четыре фазы. В фазе I устанавливают переносимость и терапевтическое действие исследуемого препара та на ограниченном числе больных (5-10 чел.), а также и на здоровых добровольцах. В фазу II клинические испытания проводят как на группе больных (100-200 чел.), так и на контрольной группе. Для получения достоверных данных применяют «двойной слепой» метод, когда ни больной, ни врач, а только руководитель ис пытания знает, какой используется препарат. Эффективность и переносимость нового фармакологического препарата сравнивают с таковыми плацебо или препаратом аналогичного действия. Фаза IV исследований изучает действие лекарственного средства на практике в разнообразных ситуациях, при этом особое внимание обращается на сбор и анализ данных о побочном действии исследуемых лекарственных препаратов. Для более эффективного использования бюджетных средств, выделяемых на здравоохранение, в настоящее время необходимо реформирование ценовой политики, что приведет к более широкому применению недорогих воспроизведенных ЛС (генериков). Рост рынка генериков в Европе, безусловно, оказывает влияние на развитие европейской фармацевтической индустрии. Замещение оригинальных ЛС аналогами стало популярной мерой правительств многих стран по сокращению затрат на лекарственное обеспечение. Генерик (в переводе с английского – калька, родовой или клан) соответс твует генеалогическому происхождению этих препаратов от родоначальника. Они производятся по аналогичной или такой же технологии, из того же исходного сырья, но чаще всего выпускаются под названием действующего химического вещества. Как правило, это точные химические копии оригинальных ЛС как по своему составу и технологии изготовления, так и по компонентам, нередко поступающим от одного и того же производителя. Генерики должны иметь такое же качество, обладать таким же лечебным эффектом, но их стоимость значительно ниже, поскольку при этом исключаются затраты, связанные с научными разработками и использованием торговых марок. Основное представление о безопасности и эффективности создаваемых оригинальных ЛС разработчики получают входе многофазовой клинической апробации, которая следует за широким доклиническим исследованием нового препарата. При выявлении каких-либо непредвиденных опасных побочных эффектов на любой стадии разработки исследования необходимо прекратить. Таким образом, эффективность и безопасность ЛС многократно проверяют в ходе разработки и, следовательно, они имеют довольно высокую степень надежности. Гарантией безопасности воспроизведенного препарата прежде всего является фармацевтическая и биологическая эквивалентность оригинальному ЛС. Фармацевтическая эквивалентность означает, что сравниваемые препараты содержат равные количества действующего вещества и соответствуют стандар там GMP. Фармацевтическую эквивалентность оценивают хорошо известными методами количественного и качественного анализа состава препарата. При этом допускаются отличия генерика от оригинального ЛС по цвету, виду лекарственной формы, упаковке. Задачи фармацевтической разработки: 1. Определение оптимального компонентного состава ЛС; 2.Обоснование спецификацией качества действующих и вспомогательных веществ с включением в них показателей качества, которые могут повлиять на эффективность и безопасность готового ЛС; 3. Разработка лекарственной формы; 4. Обоснование выбора и оптимизации технологического процесса; 5.Обоснование показателей качества, которые будут включены в спецификации при выпуске и в течение срока годности готового ЛС; 6.Обоснование выбора метода оценки эффективности и безопасности воспроизводимых ЛС. Результаты исследований по фармацевтической разработке должны убедительно гарантировать высокую степень вероятности того, что каждая единица каждой серии ЛС, производимого как для исследований, так и в условиях серийного промышленного производства, будет иметь качество, соответствующее его предполагаемому применению. На основании результатов исследований по фармацевтической разработке производитель принимает решение о разработке спецификаций и утверждений поставщиков действующих и вспомогательных веществ, производственной рецептуре, технологическом процессе, спецификации готового ЛС, сроках и условиях хранения готового ЛС. Фармацевтическая разработка представляет обоснование для выбора метода оценки эффективности и безопасности ЛС. ГФ РК занимает центральное место в системе стандартизации лекарственных средств. Фармакопея устанавливает предельный допустимый уровень качества лекарственных средств, гарантируемый государ ством. Уровень качества, регламентируемый спецификациями производителей, должен быть не ниже, но даже жестче фармакопейных требований. На основании спецификации производителя по согласованию с уполномоченным органом составляется нормативный документ по качеству лекарственного средства, предназначенный для контроля качества на всех стадиях его жизненного цикла. Таким образом, требования фармакопеи охватывают полный жизненный цикл лекарственных средств. Подобный механизм функционирования определяет роль ГФ РК как главного инструмента государственного регулирования качества лекарственных средств (рисунок 1). 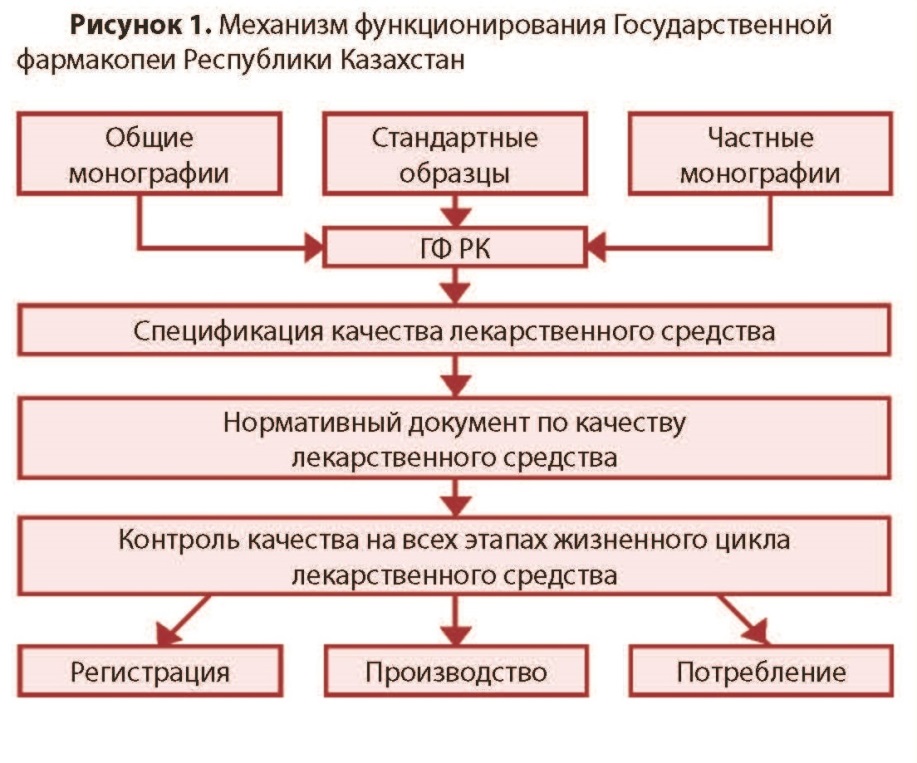 Фармакопея неразрывно связана с надлежащими фармацевтическими практиками (GхP). Надлежащие фармацевтические практики призваны обеспечивать соответствие требованиям фармакопеи от серии к серии для каждой единицы лекарственной формы. С другой стороны, необходимость выполнения требований фармакопеи стимулирует внедрение и постоянное поддерживание GхP в сфере обращения лекарственных средств. Наряду с надлежащими фармацевтическими практиками ГФ РК является важнейшим фактором обеспечения конкурентоспособности и экспортоориентированности отечественной фармпродукции. В определяющей степени этому способствует и гармонизация ГФ РК с основными фармакопеями мира. Гармонизация со стандартами основных фармакопей мира является главным принципом, положенным в основу создания ГФ РК. Для приобретения опыта разработки национальных фармакопейных стандартов республикой осуществлен ряд важных шагов по вступлению в ведущие фармакопейные организации мира, например, в Европейскую фармакопейную комиссию Совета Европы в качестве страны-наблюдателя (июнь 2006 года), в Фармакопейную конвенцию США сначала в качестве страны-наблюдателя (июль 2009 года), а затем полноправного члена (ноябрь 2010 года). Статус страны-наблюдателя или члена в ведущих фармакопейных организациях мира, помимо опыта фармакопейной стандартизации, позволяет решать следующие задачи: - определение национальных подходов и путей развития в данной области; - возможность гармонизации с требованиями основных фармакопей мира; - привлечение к работе экспертных групп; - участие в образовательных или научных программах; - аккредитация испытательных лабораторий и вступление их в сеть официальных лабораторий контроля качества лекарственных средств Совета Европы (OMCLGEON); - включение в глобальную фармакопейную деятельность (например, создание Глобального фармакопейного индекса, участие в подготовке руководства ВОЗ «Надлежащая фармакопейная практика» (GPhP) и др.); - позиционирование ГФ РК как национальной фармакопеи независимого государства. Обязательным условием гармонизации с основными фармакопеями мира является соблюдение авторских прав их патентообладателей, что требует официального разрешения на гармонизацию фармакопейных текстов. В соответствии с данным условием получено разрешение Европейского директората по контролю качества лекарственных средств Совета Европы (EDQM) в 2007 году, заключены Соглашения с Фармакопейной конвенцией США (USP) в 2010 году и Агентством Великобритании по регулированию обращения лекарственных средств и продуктов здравоохранения (MHRA) в 2014 году. Таким образом, в настоящее время ГФ РК гармонизирована с тремя основными фармакопеями мира – Европейской фармакопеей, Британской фармакопеей и Фармакопеей США [2-4]. Применение стандартов действующих в республике фармакопей осуществляется по принципу их приоритетности. Первый уровень приоритетности отводится ГФ РК как главному национальному стандарту качества лекарственных средств. При отсутствии в ней отдельных монографий или в случае их поздней актуализации применяются соответствующие монографии других фармакопей, с которыми гармонизирована ГФ РК (рисунок 2).  В настоящее время ГФ РК включает 191 общий раздел, 24 общих текста и 90 общих монографий (рисунок 3). Изложенные в них общие требования распространяются на следующее: - испытания и методы испытаний; - реактивы; - показатели качества; - упаковочные материалы и контейнеры; - субстанции для фармацевтического применения; - лекарственные формы; - лекарственные препараты, в том числе гомеопатические препараты, радиофармацевтические препараты; - биологические продукты; - лекарственные растительные средства; - морфологические группы лекарственного растительного сырья; - изделия медицинского назначения (катетеры внутрисосудистые, шовные материалы). 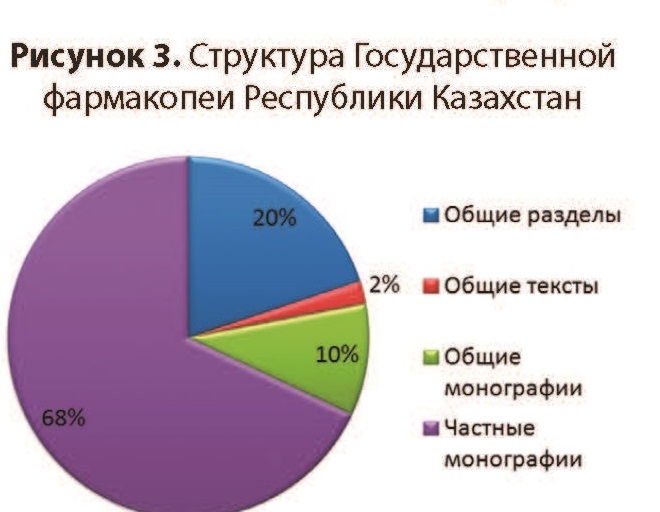 Наряду с общими монографиями ГФ РК содержит 646 частных монографий, регламентирующих требования к показателям качества, методикам испытаний и критериям их приемлемости (рисунок 4). Частные монографии распространяются на следующее: - субстанции для фармацевтического применения, в том числе активные субстанции и вспомогательные вещества; - лекарственные препараты в различных лекарственных формах; - лекарственное растительное сырье и лекарственные растительные препараты; - радиофармацевтические препараты и исходные материалы для радиофармацевтических препаратов; - медицинские иммунобиологические препараты (вакцины, иммуноглобулины). Основными пользователями фармакопеи являются разработчики, производители и дистрибьюторы лекарственных средств, аптечные организации, лаборатории контроля качества, регуляторные и экспертные органы, научные и образовательные учреждения. 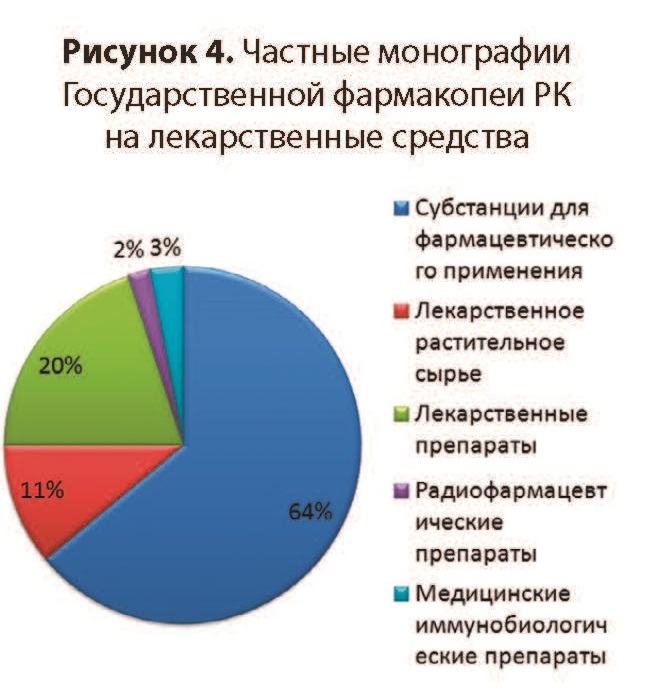 Требования ГФ РК обязательны для всех предприятий и орга низаций Республики Казахстан, занимающихся производством, изготовлением, реализацией, хранением, экспертизой, регистрацией, кон тролем и применением лекарственных средств. Создание ГФ РК имеет важное социальное, экономическое и научное значение, так как благодаря установленным стандартам, фармакопея является: - главным инструментом государственного регулирования качества лекарственных средств и изделий медицинского назначения на рынке республики; - основой экспертизы и контроля качества лекарственных средств и изделий медицинского назначения для регуляторных и экспертных органов; - фактором, определяющим конкурентоспособность и экспортоориентированность отечественной фармацевтической продукции; - эффективным инструментом защиты рынка от недоброкачественной и фальсифицированной продукции; - фактором, стимулирующим развитие научных исследований по разработке лекарственных средств и изделий медицинского назначения; - учебным и справочным пособием при подготовке фармацевтов и химиков в высших и средних специальных учебных заведениях. Создание ГФ РК является важным достижением независимого Казахстана, проявлением заботы государства о здоровье общества. Целью Государственной Фармакопеи Республики Казахстан является создание единообразного подхода в координации и совершенствовании контроля качества лекарственных средств на территории Республики Казахстан, путем утверждения единых стандартов, гармонизированных с международными, в частности с Европейской Фармакопеей. Государственная Фармакопея - это фундамент системы обеспечения безопасности и качества фармацевтической продукции на рынке Казахстана. Сегодня выполнен только первый этап огромной работы по стандартизации лекарственных средств в Казахстане, намечены реальные шаги и меры по скорейшей реализации данного проекта. Государственная Фармакопея - это один из инструментов защиты рынка от продукции сомнительного качества и различного рода фальсификаций, и как следствие - обеспечение высокого качества лекарственных средств, поступающих на отечественный рынок. Работа над Государственной Фармакопеей Республики Казахстан осуществлялась научным коллективом РГП «Национальный центр экспертизы лекарственных средств» при поддержке Министерства здравоохранения Республики Казахстан, Комитета фармацевтического контроля, с участием ряда научно-исследовательских институтов и лабораторий Российской Федерации, Украины, республиканских ведомств, отечественных предприятий фармацевтической промышленности. Фармакопеи Республики Казахстан - это только начало большого пути по обеспечению государственной политики в области качества лекарств. И весьма важным моментом на этом пути является ее целевое применение всеми участниками фармацевтического рынка в контексте обеспечения качества, эффективности и безопасности лекарственных средств на благо здоровья населения. Создание ГФ PK является важным достижением суверенного Казахстана, проявлением заботы государства о здоровье населения. Оно имеет важное социальное, экономическое и научное значение, так как благодаря установленным стандартам: позволит обеспечить высокое качество лекарственных средств, поступающих на фармацевтический рынок республики; явится одним из инструментов защиты фармацевтического рынка страны от продукции сомнительного качества и различного рода фальсификаций; явится фактором, стимулирующим внедрение требований надлежащей производственной практики в отечественную фармацевтическую промышленность; будет способствовать развитию научных исследований по созданию оригинальных лекарственных средств, в том числе из отечественного сырья. Первое издание ГФ PK подготовлено Республиканским государственным предприятием «Национальный центр экспертизы лекарственных средств, изделий медицинского назначения и медицинской техники» Министерства здравоохранения Республики Казахстан. Процесс создания ГФ PK был начат с определения концепции, принципов и подходов, положенных в ее основу. В качестве главного ориентира при создании ГФ PK были приняты требования Европейской Фармакопеи. Создание ГФ PK охватывало этапы разработки, трехкратного рецензирования, перевода на государственный язык и тщательного редактирования фармакопейных статей. Корректность частных фармакопейных статей (монографий) на готовые лекарственные средства определялась путем валидации методик испытаний и последующей их апробации в аккредитованных испытательных лабораториях. Перечень лекарственных средств, требующих разработки монографий, был определен на основе следующих факторов: > значительная доля присутствия лекарственных средств на фармацевтическом рынке республики; Ir многочисленность торговых наименований одного лекарственного средства; г перечень лекарственных средств для лечения социально-значимых заболеваний. Монографии, относящиеся к воспроизведенным (генерическим) лекарственным препаратам, были отработаны на основе сравнительного анализа их качества и установления фармакопейного уровня требований к ним, проведенных с высокой степенью скрупулезности. Каждая статья представляет собой результат острых профессиональных дискуссий на заседаниях Фармакопейной комиссии, а также широкого обсуждения фармацевтической общественностью. Государственная услуга – «Государственная регистрация, перерегистрация и внесение изменений в регистрационное досье лекарственных средств, изделий медицинского назначения и медицинской техники» (далее – государственная услуга). 2. Стандарт государственной услуги разработан Министерством здравоохранения Республики Казахстан.
Экспертиза лекарственного средства при государственной регистрации, перерегистрации и внесении изменений в регистрационное досье в соответствии с приказом МЗСР РК от 18 ноября 2009 года № 735 «Об утверждении Правил государственной регистрации, перерегистрации и внесения изменений в регистрационное досье ЛС, ИМН и МТ», состоит из следующих этапов:
Перечень документов при проведении экспертизы ЛС:
Качество лекарственных средствопределяется как соответствие лекарственных средств государственному стандарту их качества. Доку ментом, подтверждающим соответствие качества лекарственного сред ства государственному стандарту, является сертификат качества лекар ственного средства. Безопасность означает характеристику лекарственных средств, ос нованную на сравнительном анализе их эффективности и оценке риска причинения вреда здоровью. Эффективность лекарственных средств, в свою очередь, означает характеристику степени положительного влия ния на течение болезни. Качество лекарственных средств определяется как соответствие лекарственных средств государственному стандарту их качества. Доку ментом, подтверждающим соответствие качества лекарственного сред ства государственному стандарту, является сертификат качества лекар ственного средства. Безопасность означает характеристику лекарственных средств, ос нованную на сравнительном анализе их эффективности и оценке риска причинения вреда здоровью. Эффективность лекарственных средств, в свою очередь, означает характеристику степени положительного влия ния на течение болезни. безопасность лекарственных средств - отсутствие недопустимого риска, связанного с возможностью нанесения вреда здоровью человека; эффективность лекарственного средства - характеристика степени влияния лекарственного средства на клинические проявления (патологическое состояние) и причины заболевания; Система обеспечения качества и безопасности лекарственных средств в Казахстане. на протяжении всего его жизненного цикла – от разработки до конечного потребителя, и весь этот процесс регламентирован законодательством Республики Казахстан. Если проследить путь движения препарата в системе государственного контроля, то можно увидеть, что без контроля не остаются ни научные исследования физико-химических свойств молекулы, ни обширные доклинические и клинические исследования. Все это отслеживается в Казахстане во время проведения обязательной процедуры государственной регистрации в контролирующем органе – Комитете контроля медицинской и фармацевтической деятельности Министерства здравоохранения Республики Казахстан. Без регистрационного удостоверения о государственной регистрации лекарственного средства, полученного в установленном порядке, препарат просто не может попасть в обращение. Именно процедура государственной регистрации является мощным барьером от появления на нашем рынке недоброкачественных и небезопасных препаратов, при проведении которой подтверждается качество, безопасность и, конечно же, действенность лекарственного средства. При государственной регистрации проводится полная экспертиза лекарственного средства, включающая первичную экспертизу документов и материалов предоставляемых фармацевтической фирмой, аналитическую экспертизу лекарственного средства на соответствие требованиям нормативно-технического документа по контролю за качеством и безопасностью лекарственного средства, специализированную фармацевтическую и фармакологическую экспертизы. Каждый последующий этап экспертизы лекарственного средства проводится на основании положительного заключения предыдущего этапа экспертизы. Так, благодаря этой процедуре в 2010 году не прошли государственную регистрацию и не допущены на фармацевтический рынок Республики Казахстан 512 наименований лекарственных средств, в 2011 году – 715, в 2012 году – 366, а в 2013 году – 292. Основными причинами отказа в государственной регистрации заявленных препаратов были такие причины, например, как: более низкая безопасность и эффективность препарата по сравнению с ранее зарегистрированными аналогами; несоответствие установленным показателям качества и безопасности; получение отрицательных результатов клинических и (или) других исследований; Факты: не прошли государственную регистрацию и не допущены на фармацевтический рынок Республики Казахстан - в 2010 году 512 наименований лекарственных средств, в 2011 году – 715, в 2012 году – 366, в 2013 году – 292. наличие в составе препарата веществ, запрещенных к применению в Республике Казахстан; наличие в регистрационном досье недостоверных сведений и др. Подтверждение безопасности, качества и эффективности лекарства при проведении процедуры государственной регистрации основывается на сравнительном анализе его эффективности и оценке риска причинения вреда здоровью. Всю вышеперечисленную экспертизу при государственной регистрации лекарственных средств проводит специально созданная государственная экспертная организация – РГП «Национальный центр экспертизы лекарственных средств, изделий медицинского назначения и медицинской техники» (НЦЭЛС) Министерства здравоохранения РК, которая является монополистом по своему роду деятельности и не принимает непосредственного участия в разработке\производстве лекарственного средства, и поэтому может непредвзято осуществлять эту деятельность. Сегодня лаборатории НЦЭЛС, в состав которых входит Республиканская иммунобиологическая лаборатория, оснащены самым современным оборудованием, позволяющим воспроизводить все сложные и необходимые методики качественного анализа. В настоящее время проводятся подготовительные мероприятия по вступлению НЦЭЛС в Единую сеть официальных испытательных лабораторий по контролю качества лекарственных средств при Европейской фармакопее, продолжается работа по созданию III тома национального фармакопейного стандарта – Государственной фармакопеи Республики Казахстан (ГФ РК)- свода стандартов, гармонизированных с международными требованиями, являющегося своего рода основой для обеспечения качества и безопасности лекарств. Свидетельством высоких достижений в области совершенствования стандартов качества и безопасности лекарств при разработке отечественной ГФ РК, служит тот факт, что Казахстан является официальным наблюдателем Европейской фармакопейной комиссии Совета Европы, членом Фармакопейной конвенции США с предоставлением права решающего голоса, подготовлен к подписи договор о совместном сотрудничестве с Британской Фармакопеей. После проведения государственной регистрации лекарственное средство может быть произведено или ввезено на территорию Республики Казахстан. После чего начинается Лекарственный информационно Выпуск 6 Стр 2 -аналитический центр Бесплатный номер по Казахстану 8 800 080 88 87 WWW.DRUGINFO.KZ Система обеспечения качества и безопасности лекарственных средств в Казахстане. следующий, немаловажный этап, обеспечивающий контроль каждой серии произведенного или ввезенного препарата – оценка безопасности и качества. Важно, что в настоящее время лекарственные средства вышли из системы технического регулирования, ввиду своей узкой специфичности, применения более жестких обязательных требований, нежели к другой продукции, такой, например, как пищевая или товары легкой промышленности. Оценке безопасности и качества подвергаются все лекарственные средства при их ввозе, производстве, а также она имеет место при возникновении сомнения в качестве лекарственных средств, зарегистрированных в Республике Казахстан, в том числе изъятых государственным органом, для подтверждения факта обнаружения фальсифицированных лекарственных средств. Более подробную информацию о порядке проведения процедуры оценки безопасности и качества лекарственных средств можно получить, изучив Правила проведения оценки безопасности и качества лекарственных средств, утвержденные постановлением Правительства Республики Казахстан от 14 декабря 2012 года №1606. В цепочке государственных процедур, кроме государственной регистрации и по серийной оценки безопасности и качества лекарственных средств, немаловажную роль играет лицензирование фармацевтической деятельности, которое призвано обеспечить необходимые условия для сохранения качества лекарственных средств при их производстве, хранении, транспортировке и реализации. На этапе применения лекарственного средства пациентом немаловажную роль приобретают вопросы правильного хранения и применения препарата. С целью содействия рациональному назначению лекарственных средств, эффективному и безопасному их применению создана специализированная государственная организация, предоставляющая достоверную и полную информацию о лекарственных средствах – Лекарственный информационно-аналитический центр, который имеет сеть региональных отделов во всех регионах страны. Именно от знания пациентом вопросов рационального применения и надлежащего хранения препаратов в домашних условиях, зависит качество, эффективность и, отчасти, безопасность назначаемой врачом фармакотерапии. Лекарственным центром ежегодно выпускаются справочники, основанные на достоверной информации из лучших Все ведущие международные эксперты сошлись во мнении о том, что качество и безопасность лекарственных средств могут быть обеспечены только строгим соблюдением правил Надлежащей производственной практики (GMP), Надлежащей клинической практики (GCP), Надлежащей лабораторной практики (GLP), Надлежащей дистрибьюторской практики (GDP), Надлежащей аптечной практики (GPP), Надлежащей регуляторной практики (GRP). международных источников доказательной медицины, методические рекомендации и лекарственные бюллетени, содержащие ответы на актуальные вопросы практического здравоохранения. Нужно подчеркнуть, что сфера обращения лекарственных средств претерпевает ряд существенных и прогрессивных изменений. Министерством здравоохранения Республики Казахстан проводится активная работа по переносу акцента над непосредственным контролем за качеством лекарственных средств, к более совершенной системе по его обеспечению. В большинстве развитых стран действует именно такая модель, стандартизирующая каждый элемент обеспечения качества лекарственного средства и направленная именно на предупреждение случаев появления дефектов качества, а не на его устранение. Все ведущие международные эксперты сошлись во мнении о том, что качество и безопасность лекарственных средств могут быть обеспечены только строгим соблюдением правил Надлежащей производственной практики (GMP), Надлежащей клинической практики (GCP), Надлежащей лабораторной практики (GLP), Надлежащей дистрибьюторской практики (GDP), Надлежащей аптечной практики (GPP), Надлежащей регуляторной практики (GRP). Идеология поэтапного перехода казахстанской фармацевтической отрасли на стандарты надлежащих практик закреплена в основополагающем стратегическом документе – Государственной программе развития здравоохранения Республики Казахстан «Саламатты Казахстан». Кроме того, создан необходимый правовой фундамент для внедрения стандартов в Кодексе Республики Казахстан «О здоровье народа и системе здравоохранения». Следует отметить, что необходимость перехода на стандарты международных практик принята и поддерживается фармацевтической общественностью республики и уже 4 отечественных производственных участка добровольно перешли на стандарт GMP, 11 объектов оптовой реализации лекарственных средств получили заключения о соответствии стандарту GDP и 15 объектов розничной реализации лекарственных средств получили заключения о соответствии стандарту GPP. В настоящее время в Республике Казахстан уже подготовлен фармацевтический инспекторат и ведется активная подготовительная работа по внедрению стандартов, в том числе разработке механизмов стимулирования объектов фармацевтической деятельности к переходу на стандарты обеспечения качества и безопасности лекарств. |