Хроматографические методы анализа и их использование в анализе объектов окружающей природной среды
 Скачать 330.89 Kb. Скачать 330.89 Kb.
|

|
Глава 3. Примеры применения хроматографии в анализе объектов окружающей среды Анализ состояния водной среды с помощью метода газовой хроматографии[13-15] Метод газовой хроматографии для анализа состояния водной среды все шире проникает из области научного эксперимента в сферы, непосредственно связанные со многими сторонами жизни человека. В большинстве случаев термин "водная среда" относят к различным водным объектам, находящимся вне организма человека - к морским и речным водам, иным поверхностным водам суши, подземным водам, промышленным и бытовым стокам, атмосферным осадкам, наконец, к пищевым водам (схема 1). Все эти группы объектов было бы правильнее называть "внешней водной средой". Схема 1 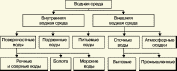 В связи с тем, что в организме человека, как и многих других живых существ, по крайней мере 80% приходится на долю воды, правомерно говорить о "внутренней водной среде" и о соответствующих объектах анализа. Эта категория объектов включает гомогенизаты и экстракты тканей различных органов и биологические жидкости, к числу которых относятся плазма крови, лимфа, слюна, моча, желчь, желудочный сок, спинно-мозговая жидкость и другие (схема 2). Как в первой, так и во второй группе объектов в анализируемых пробах могут присутствовать весьма разнообразные вещества, детальный анализ которых требует применения различных аналитических методов. Так, например, растворенные газы в морских и подземных водах, а также и в крови человека, определяют с помощью спектральных методов и в ряде случаев с помощью газовой хроматографии. Металлы, присутствующие в водных объектах, анализируют с помощью атомной абсорбционной спектроскопии или эмиссионной спектроскопии с индуктивно связанной плазмой. Очень малые следовые примеси многих элементов в водной среде определяют с помощью радиоактивационного анализа, а состав присутствующих в водной среде анионных составляющих анализируют методом ионной хроматографии. Схема 2 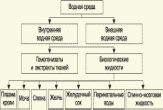 Высокомолекулярные полимерные вещества, присутствующие в водной пробе (гуминовые и фульвиновые кислоты во внешних природных водах, белковые компоненты и нуклеиновые кислоты в плазме крови), определяют методами жидкостной хроматографии - эксклюзионной, тонкослойной и высокоэффективной жидкостной хроматографии (схема 3). Схема 3 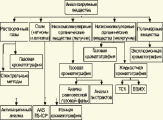 Однако очень большое число низкомолекулярных летучих органических соединений, способных переходить в парообразное состояние без разложения, анализируют и определяют количественно с помощью метода газовой хроматографии (схема 4). При этом собственно газохроматографическому определению может предшествовать операция извлечения анализируемых компонентов из водной пробы, их концентрирование и в ряде случаев перевод в их производные, обладающие более высокой летучестью либо меньшей полярностью, чем исходные соединения, и потому более пригодные для анализа с помощью газовой хроматографии (например, органические кислоты обычно переводят в их метиловые эфиры, аминокислоты - в алкиловые эфиры N-трифторацетильных производных и т.п.). Таким образом оказывается возможным использовать метод газовой хроматографии для определения тех органических веществ, которые в принципе не переходят в пар без разложения вследствие своей высокой полярности или малой термической устойчивости (например, углеводы). Схема 4 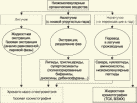 Метод газовой хроматографии уже в течение достаточно длительного времени является общепризнанным способом анализа летучих органических компонентов и следовых примесей водной среды. Опубликованы многочисленные монографии и оригинальные статьи, описывающие особенности применения метода газовой хроматографии к анализу тех или иных водных объектов. Как и любой другой аналитический метод, газовая хроматография может дать корректную объективную информацию о составе водных объектов при условии достаточно правильного выполнения предварительных операций по отбору представительных проб анализируемых вод, извлечения и концентрирования подлежащих определению компонентов и их групп и ввода их в хроматографическую систему. Объектами газохроматографического определения в водных средах могут быть растворенные газы и органические соединения с молекулярной массой от 16-30 (метан, этан) до 400-500 и более. Состав примесей может довольно быстро изменяться вследствие целого ряда причин. Поэтому время от момента отбора проб до выполнения анализа или предшествующих ему операций, обеспечивающих консервацию их состава, должно быть по возможности малым. Причины изменения состава примесей водной среды, определяемых с помощью газовой хроматографии, могут включать следующие процессы: а) потери растворенных газов и наиболее легколетучих органических компонентов вследствие изменений температуры и давления (например, при нагреве водной пробы от исходной температуры водоисточника до температуры лабораторного помещения); б) исчезновение некоторых подлежащих определению примесных компонентов водных проб в результате химических и микробиологических процессов при хранении до проведения анализа (окисление альдегидов и тиолов, гидролиз ацеталей и галогенопроизводных, микробиологическое расщепление углеводородов нефти и др.); в) загрязнение проб примесями, извлекаемыми из полимерной тары, используемой при отборе проб и их последующей транспортировке и хранении (пластификаторы, низкомолекулярные компоненты полимеров и т.п.); г) загрязнение проб примесями, содержащимися в применяемых для экстракции растворителях; д) изменение проб в испарителях и колонках применяемых хроматографических систем. Правильно организованный газохроматографический анализ должен в возможно более полной степени исключить все перечисленные выше причины изменения состава анализируемых проб. Этой цели служат многочисленные методики анализа, опубликованные в оригинальных работах, а в ряде случаев и включенные в нормативные документы. Для извлечения определяемых компонентов из водных матриц применяют методы экстракции малыми объемами органических растворителей с последующим концентрированием путем отгонки экстрагента (рис. 1); твердофазной экстракции с сорбцией определяемых компонентов на адсорбентах с привитой органической неподвижной фазой (углеводородными радикалами от С2H5 до С20Н41, либо функционально замещенными фрагментами с нитрильными, аминными или диольными группами). Разработаны методы микроэкстракции на единичном стеклянном или кварцевом волокне, покрытом пленкой полисилоксановой неподвижной фазы. 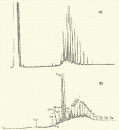 Рис. 1. Типичные хроматограммы нефтяных загрязнений, экстрагированных из воды Таганрогского залива: а) - август 1991 г.; б) - октябрь 1991 г. В пробу анализируемой воды объемом около 100 мл (в конической колбе) помещают магнитную мешалку в форме стеклянной трубки длиной 30 мм и диаметром 2-3 мм с запаянными концами. Внутрь трубки помещают стальной стержень длиной 25 мм и диаметром 1-1,5 мм, а снаружи на трубку надевают отрезок трубки из силиконовой резины длиной 25-30 мм с внутренним диаметром 1 мм и толщиной стенок 0,5 мм. Согласно опубликованным данным перемешивание водной пробы такой мешалкой со скоростью несколько сотен оборотов в минуту в течение 10-15 мин при 20 °С приводит к тому, что более 90% всех липофильных примесей абсорбируется в силиконовой оболочке мешалки. После этого мешалку можно поместить в нагретый испаритель хроматографа для немедленного проведения анализа либо сохранить ее длительное время в закрытой пробирке для транспортировки в стационарную лабораторию. Подобная техника извлечения и концентрирования малых примесей, названная авторами Stir Bar Sorptive Extraction, несомненно, имеет серьезные перспективы широкого применения. Возможно, что использование таких магнитных мешалок с полимерными покрытиями разных типов позволит избирательно извлекать из водных проб различные группы соединений, отличающиеся по своей полярности и прочим физико-химическим характеристикам. Для определения состава легколетучих компонентов водных проб оказывается плодотворным метод газохроматографического анализа равновесного пара и газовой экстракции с улавливанием извлекаемых веществ в сорбционных концентраторах и последующим криогенным вводом в капиллярную колонку. Таким способом, например, подробно изучен состав хлорсодержащих микропримесей, образующихся при хлорировании питьевой воды (рис. 2). При извлечении микропримесей полярных веществ, хорошо растворимых в воде, применяют метод экстракции полярными водорастворимыми экстрагентами (спиртом, ацетоном) с предварительным насыщением водных проб неорганическими солями (высаливание хлоридом натрия или сульфатом аммония). 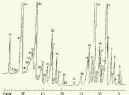 Рис. 2. Хроматограмма летучих органических соединений, содержащихся в пробе хлорированной питьевой воды. Капиллярная колонка длиной 50 м и диаметром 0,25 мм с силиконовой смазкой "Эдвардс" в качестве неподвижной фазы. Пики, обозначенные цифрами, идентифицированы. Пик 9 – хлороформ Собственно газохроматографический анализ в большинстве случаев в настоящее время осуществляют с применением высокоэффективных капиллярных колонок в условиях программирования температуры от 50-100 °С до 300-350 °С (и даже до 400 °С и выше). Скорость нагрева колонок при этом может изменяться от 3-5 до 20-30 град/мин в зависимости от характера разделяемых компонентов. Идентификацию пиков на хроматограммах выполняют с применением известных хроматографических зависимостей индексов удерживания от температур кипения и от молекулярной массы веществ. Для той же цели применяют селективные детекторы (например, электронно-захватный), термоионные хемилюминесцентные, атомно-абсорбционные, масс-селективные и др. Наиболее полную информацию о молекулярном строении определяемых веществ позволяют получить методы хромато-масс-спектрометрии и газовой хроматографии с ИК-Фурье спектроскопическим детектором. Особенно плодотворным оказалось использование этих двух методов при определении пестицидов, полихлорированных бифенилов, диоксинов и им подобных веществ, продуктов распада токсичных ракетных топлив и боевых отравляющих веществ в объектах окружающей среды (рис. 3). 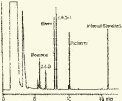 Рис. 3. Хроматограмма воды реки Оук Крик (США), содержащей пестициды: 1 - Дикамба (4,7 мкг/л); 2 - 2,4-D (7,0 мкг/л); 3 - Силвекс (4,5 мкг/л); 4 - 2,4,5-Т (5,2 мкг/л); 5 - Пиклорам (3,3 мкг/л); 6 - внутренний стандарт В ряде случаев применение селективных детектирующих систем позволяет не только повысить чувствительность определения целевых компонентов, но и выявить источники загрязнения водной среды. Так, например, применение хемилюминесцентного озонового детектора с высокой специфической чувствительностью к веществам, содержащим серу, позволяет зафиксировать профиль серосодержащих соединений нефти и нефтяных топлив. Эти компоненты значительно более устойчивы в водной среде, чем углеводородные составляющие нефтепродуктов, поэтому совпадение таких профилей позволяет с большой степенью достоверности указать источник нефтяного загрязнения данного водного бассейна (рис. 4), . 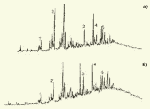 Рис. 4. Хроматограммы: а) - серосодержащих компонентов нефтяного загрязнения морской воды (район г. Таганрога); б) - судового дизельного топлива. Кварцевая капиллярная колонка длиной 25 м и диаметром 0,25 мм с полидиметилсилоксаном SE-54 в качестве неподвижной фазы. Программирование температуры от 60 до 280 °C cо cкоростью нагрева 10 град/мин: 1 - метилбензотиофены; 2 - диметилбензотиофены; 3 - дибензотиофен; 4 - метилдибензотиофены; 5 – диметилдибензотиофены Большие возможности газохроматографического анализа в настоящее время определенно позволяют осуществить достаточно полный анализ водных проб практически любого происхождения, в том числе питьевых, речных, озерных и морских вод, сточных вод коммунальных систем и промышленных предприятий. В ряде случаев такие анализы могут быть проведены в полевых условиях непосредственно в местах отбора проб. Тем не менее, пока не существует методов, позволяющих провести полный анализ водных проб в единой системе без проведения предварительных лабораторных операций, часто трудоемких и длительных. Перспективными для создания таких методов следует считать хроматографические системы с использованием в качестве подвижных фаз парообразных и сверхкритических сред (в том числе паров воды). Широко используется газовая хроматография также и для анализа объектов внутренней водной среды. При этом применяется весь арсенал технических приемов, разработанных за 50 лет развития газовой хроматографии: высокоэффективные капиллярные колонки, высокочувствительные селективные детекторы, комбинированные аналитические системы, сочетающие газовую хроматографию с масс-спектрометрией и с ИК-спектроскопией с Фурье-преобразованием. В этой области сложились два основных направления аналитических исследований. С одной стороны, это изучение состава естественных сред организма (плазмы крови, мочи, слюны, спинно-мозговой жидкости и др.) с целью выявления изменений этого состава в зависимости от физиологических особенностей организма, наличия патологических изменений и тому подобных факторов. В настоящее время это направление газохроматографического анализа внутренней водной среды организма отражено в ряде руководств и монографий. Показано, что во многих случаях газохроматографический анализ позволяет выявить на ранней стадии целый ряд заболеваний и таким образом ускорить их лечение (рис. 5). При таких исследованиях широко используют экстракцию целевых компонентов растворителями, твердофазную экстракцию и метод анализа равновесного пара (рис. 6). 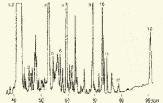 Рис. 5. Метаболический профиль стероидов из мочи пациента с опухолью яичника, секретирующей тестостерон. Идентифицированы все компоненты. Увеличенное содержание компонентов 1, 2, 4 и 7 указывает на наличие опухоли (1 - андростерон; 2 - этиохоланон; 4 - 11b-оксиандростерон; 7 - прегнантриол) 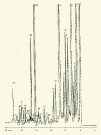 Рис 6. Хроматограмма летучих органических соединений мочи, полученная на капиллярной колонке с полисилоксановой неподвижной фазой БС-2 в условиях программирования температуры: 1 - ацетон; 3 - этанол; 6 - 2-пентанон; 7 - н-пропанол; 10 - диметилдисульфид;13 - 4-гептанон; 14 - н-бутанол; 15 - 2-гептанон; 29 - пиррол; 45 – карвон Вторым важным направлением в анализе объектов внутренней водной среды является выявление чужеродных для организма соединений (фармацевтических препаратов, разного рода ядов, алкоголя, других наркотических веществ и т.п.). Так, применение высокочувствительного термоаэрозольного детектора, избирательно регистрирующего азотсодержащие соединения, позволило регистрировать противосудорожные лекарственные средства в крови детей, больных эпилепсией, даже через 3-5 дней после их применения (рис. 7). Аналогично, с помощью селективного термоионного детектора с высокой чувствительностью регистрировали в плазме крови наличие анестезирующего препарата кетамина, находящего, к сожалению, довольно широкое применение в качестве галлюциногенного наркотика (рис. 8).  Рис. 7. Хроматограмма 0,1% раствора противоэпилептических лекарственных средств. Кварцевая капиллярная колонка длиной 10 м и диаметром 0,25 мм с полисилоксаном OV-17 в качестве неподвижной фазы: 1 - этанол; 2 - фенобарбитал; 3 - гексамидин; 4- дифенин  Рис. 8. Хроматограмма смеси компонентов плазмы крови, содержащей кетамин: а) - пламенно-ионизационный детектор; б) - термоионный детектор; 1 - кетамин; 2 - внутренний стандарт Подобным же способом при газохроматографическом анализе проб плазмы крови, мочи или слюны могут быть определены несколько сотен других наркотиков, веществ, используемых в качестве допинга в спортивных соревнованиях, и анаболических стероидов, запрещенных к употреблению международными спортивными организациями. Все перечисленные выше достижения газохроматографического метода в области анализа объектов водной среды все шире проникают из области научного эксперимента в сферы, непосредственно связанные с многими сторонами жизни современного общества. К таким областям можно отнести изучение состояния окружающей среды, контроль качества питьевой воды, сельскохозяйственной продукции и пищевых продуктов, клиническую медицину, криминалистику и ряд других. Определение полиароматических углеводородов в объектах окружающей среды методами жидкостной и тонкослойной хроматографии[14] Было определено содержание полиароматических углеводородов (ПАУ), в частности, бенз(а)пирена в снежном покрове. Пробоподготовку осуществляли экстракцией диэтиловым эфиром. Качественный анализ осуществляли методом тонкослойной хроматографии. Пробы наносили на пластину Silufol UV-254 и осуществляли хроматографический анализ в двух системах: система 1 - раствор кофеина в хлороформе; система 2 - смесь циклогексана и н-гексана. Использование системы 1 позволило снизить нижний предел обнаружения. Количественный анализ осуществляли методом газожидкостной хроматографии (ГЖХ). Исследование проводили на хроматографе «Цвет-500» с пламенно-ионизационным детектором. В качестве сорбента использовали силиконовый каучук SE-54 с нанесенной на него неподвижной фазой OV-101. Анализ проводили в режиме программирования температуры от 200 до 310 С со скоростью 4 С /мин. В качестве газа-носителя использовали азот. Метод позволил определить ПАУ на уровне ПДК. |