хроматография-лекции. Хроматографические методы. Общая характеристика методов
 Скачать 6.82 Mb. Скачать 6.82 Mb.
|

1.1. Характеристики хроматографического разделениякомпонентов анализируемой смеси Результатом хроматографического разделения исследуемой пробы является хроматограмма. Различают внутреннюю и внешнюю хроматограммы. Внутренняя хроматограмма – это распределение разделенных веществ вдоль колонки в виде отдельных полос (зон). Внешняя хроматограммма – это графическое изображение распределения веществ в элюате, кривая зависимости сигнала детектора хроматографа от времени или от объема элюата, прошедешего через колонку. (Элюат – подвижная фаза, выходящая из колонки и содержащая разделенные компоненты.) 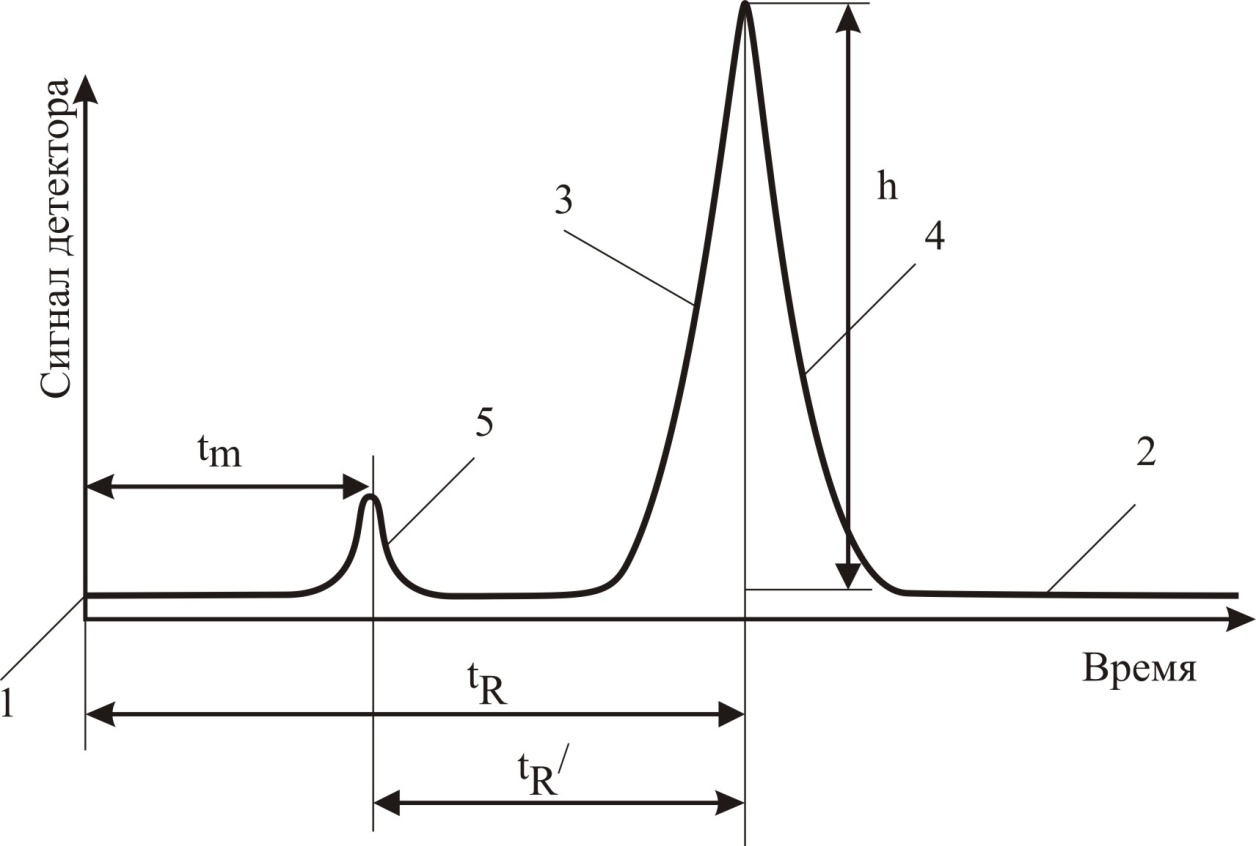 Идеализированная внешняя хроматограмма представлена на рис. 1.4. На внешней хроматограмме по оси абсцисс отложено время хроматографирования (можно отложить объем элюата), по оси ординат – аналитический сигнал детектора хроматографа, зависящий от содержания вещества в элюате и чувствительности детектора к компонентам анализируемого вещества. На дифференциальной хроматограмме различают следующие составные части: 1 –точка ввода пробы; 2 – нулевая линия, участок хроматограммы, полученной при регистрации сигнала дифференциального детектора во время выхода из колонки чистой подвижной фазы; 5 – пик несорбирующегося компонента. Пик - участок хроматограммы, полученной при регистрации сигнала детектора во время регистрации одного из компонентов (или смеси нескольких неразделенных компонентов)ограничивается фронтом (3), соответствующим возрастанию концентрации компонента до максимальной, и тылом (4), отвечающим убыванию концентрации компонента в подвижной фазе. Расширение полосы компонента по мере хроматографического разделения, ведущее к получению широкого хроматографического пика, называют размытием пика. Размытие может быть симметричным и асимметричным. В последнем случае образуется пик либо с размытым фронтом, либо с размытым тылом.  1.1.1 Первичные параметры удерживания К числу первичных параметров удерживания относятся: время удерживания, объем удерживания и соответствующий им отрезок на хроматограмме расстояние удерживания (рис.). Время от момента ввода анализируемой пробы до регистрации, максимума пика называют временем удерживания (элюирования) tR данного компонента. Время удерживания каждого компонента складывается из двух составляющих – времени пребывания в подвижной фазе tm и неподвижной фазе tR': 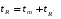 ’ (1.1) ’ (1.1)Значение tm фактически равно времени прохождения через колонку несорбируемого компонента (газа-носителя в газовой хроматографии, элюата в жидкостной.) Время удерживания tR не зависит от количества пробы, но зависит от природы вещества и сорбента, упаковки сорбента, скорости подачи подвижной фазы и может меняться от колонки к колонке. Поэтому истинную способность данного вещества удерживаться в хроматографической колонке характеризуют исправленным временем удерживания tR': 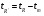 . (1.2) . (1.2)Для характеристики удерживания часто применяется понятие удерживаемого объема VR – объем подвижной фазы, который нужно пропустить через колонку с определенной скоростью, чтобы элюировать вещество: 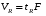 , (1.3) , (1.3)где F – объемная скорость потока подвижной фазы, см3/с. Объем для вымывания несорбируемого компонента выражается через tm: 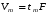 (1.4) (1.4)Соответственно исправленный удерживаемый объем равен: 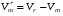 (1.5) (1.5)При постоянных условиях хроматографирования (скорость потока элюента, давление газа носителя, температура, состав фаз) значения tR и VR строго воспроизводимы и используются для идентификации веществ. Массу вещества, вымываемого из колонки, можно найти по площади под кривой элюирования: 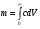 (1.6) (1.6)где с – концентрация, ммоль/мл; V – объем, мл 1.2.1. Основные закономерности сорбционных процессов. В основе хроматографического разделения лежат, прежде всего, сорбционные процессы. Как большинство физико-химических процессов они проходят две стадии: стадию приближения к равновесию, которая развертывается во времени, характеризуется определенной скоростью и изучается в разделе кинетики сорбции, и стадию собственно равновесную, закономерности которой описываются статикой сорбции. Последняя стадия играет решающую роль в достижении хроматографического разделения. Под сорбцией понимают поглощение газов, паров или растворенных веществ твердыми или жидкими поглотителями. При этом поглощаемые вещества называют сорбатами, а поглотители – сорбентами. Если при этом сорбат поглощается всем объемом сорбента, то процесс называют абсорбцией, а если он концентрируется на поверхности сорбента, то адсорбцией; соответственно и сорбенты делятся на абсорбенты и адсорбенты. Чаще всего адсорбентами являются твердые тела с развитой поверхностью, в хроматографии широко применяют для этой цели силикагели, алюмогели, активные угли, молекулярные сита, пористые полимерные сорбенты. Жидкие поглотители (абсорбенты) сами по себе в аналитической хроматографии не используют, их обычно наносят на поверхность твердых материалов с относительно небольшой поверхностью, которые называют твердыми носителями. В этом случае наряду с абсорбцией и адсорбцией на поверхности жидкого поглотителя, называемого в хроматографии неподвижной фазой, может происходить адсорбция и на поверхности твердого носителя. Таким образом, в хроматографии применяют два основных типа сорбентов: твердые адсорбенты и неподвижные фазы, нанесенные на твердый носитель. Единой стройной теории, количественно описывающей весь процесс хроматографического разделения, до настоящего времени нет. Установление теоретической зависимости между химическим строением веществ и его коэффициентом распределения между фазами, знание которого позволяет предсказать хроматографическое поведение вещества, является задачей, которая еще ждет своего решения. Поэтому в настоящее время предложен ряд теоретических подходов, использующих некоторые допущения и позволяющих достаточно удовлетворительно описывать ход хроматографического процесса. С одной стороны, хроматографический процесс можно рассматривать с точки зрения формы изотермы распределения, т.е. зависимости между концентрацией вещества в подвижной фазе и его сорбцией неподвижной фазой. В этом случае в основу описания будут положены процессы, ответственные за разделение веществ при хроматографировании. С другой стороны, хроматографический процесс можно рассматривать с точки зрения времени установления равновесия в процессе массообмена между сорбентом и поглощаемым веществом. В этом случае в основу описания будут положены процессы, влияющие на степень размывания хроматографических полос и ухудшающие разделение. В первом случае следует различать хроматографию линейную и нелинейную, а во втором – идеальную и неидеальную хроматографию. 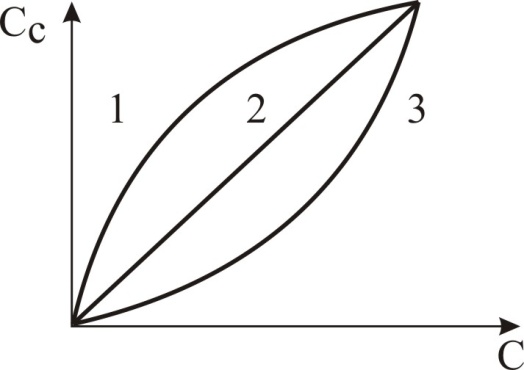 При этом линейная хроматография предполагает описание взаимодействия сорбент-сорбат линейной изотермой, а неидеальная хроматография исходит из того, что скорость установления равновесия является конечной величиной. Рассмотрим следующий процесс. Пусть имеем хроматографическую колонку, в которой на расстоянии “ x ” от верхней границы адсорбента расположен фронт зоны исследуемого соединения. При введении в колонку порции подвижной фазы в объеме “ dv ”, зона вещества смещается по колонке на расстояние dx. Изменение концентрации вещества в подвижной фазе в этих условиях будет определяться двумя основными процессами: • вещество, растворенное в подвижной фазе, фильтруется между зернами адсорбента и перемещается по колонке без образования адсорбционных связей; • вещество диффундирует в объем зерен адсорбента с образованием адсорбционных связей определенной силы. В таком случае, уравнение, описывающее суммарное изменение концентрации вещества в подвижной фазе по высоте колонки в ходе хроматографического процесса, запишется следующим образом:  Первое слагаемое в правой части уравнения (1) описывает изменение концентрации вещества при его фильтрации между зернами адсорбента без образования адсорбционных связей, второе cлагаемое – изменение концентрации вещества, обусловленное протеканием адсорбционного процесса. Коэффициент “α ” характеризует степень плотности упаковки адсорбента в колонке: долю объема пустот между зернами адсорбента. Диапазон изменения численных значений коэффициента “α ” − от нуля до единицы. В том случае, когда величина степени плотности упаковки адсорбента в колонке очень высокая, объем пустот между зернами мал, величина коэффициента “α ” стремится к нулю и изменение концентрации вещества будет обусловлено преимущественно вкладом второго слагаемого, отражающего особенности протекания процесса адсорбции. В случае очень рыхлой упаковки адсорбента в колонке величина коэффициента “α” стремится к единице и роль вклада первого слагаемого в величину суммарного изменения концентрации вещества в подвижной фазе существенно увеличивается. В пределе, если адсорбент в колонке отсутствует, второе слагаемое в уравнении исчезает и изменение концентрации вещества описывается только процессами, происходящими в подвижной фазе. Умножим обе половины равенства на dv/dC, получим:  Разделив единицу на обе части равенства (2), получим:  Из полученного уравнения вытекает интересное следствие: левая часть уравнения характеризует величину скорости перемещения фронта зоны по мере прибавления новых порций подвижной фазы в колонку. Видно, что величина скорости перемещения фронта зоны зависит от величины производной dm/dC и, следовательно, обусловлена параметрами изотермы адсорбции, поскольку эта функциональная зависимость и есть изотерма адсорбции. Таким образом, приходим к весьма важному выводу – скорость перемещения фронта зоны определяется параметрами изотермы адсорбции. Из закона Генри, описывающего начальный линейный участок изотермы распределения m=f(С) следует: 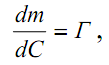 где Г – константа изотермы распределения Генри. Подставив это выражение в уравнение (3), получим:  Таким образом, скорость перемещения данной концентрации компонента в подвижной фазе вдоль колонки зависит от константы изотермы распределения Генри. Из уравнения (4) следует, что эта скорость будет тем больше, чем меньше константа Генри, т.е. чем хуже адсорбируется данный компонент. И, наоборот, скорость будет тем меньше, чем сильнее этот компонент адсорбируется. Поэтому хроматографические полосы, соответствующие разным компонентам, перемещаются вдоль колонки с постоянными, но разными скоростями, что и обеспечивает разделение этих компонентов. Поскольку каждая концентрация исследуемого соединения “C ” в подвижной фазе передвигается вдоль колонки с постоянной скоростью “u ”, то распределение С=f(x), создавшееся у входа в колонку при вводе пробы, переместится к выходу из колонки без изменения, и хроматографическая полоса соответствующего компонента не будет размыта. Такое положение характерно только для линейной идеальной хроматографии. Рассмотренный подход предполагает, что равновесное распределение компонента между фазами устанавливается мгновенно. Однако в реальном хроматографическом процессе оно устанавливается за определенное время и поэтому хроматографическая полоса при движении вдоль колонки размывается. Это происходит вследствие ряда динамических и кинетических причин. Во−первых, в насадочных хроматографических колонках, сказывается диффузия молекул адсорбирующегося соединения вдоль и навстречу потоку подвижной фазы (продольная диффузия), перенос и диффузия молекул вокруг зерен адсорбента (вихревая диффузия), а также диффузия молекул в поры адсорбента (внутренняя диффузия). Во−вторых, молекулы компонента, находясь в неподвижной фазе, отстают от таких же молекул, переносимых подвижной фазой, вследствие конечной скорости процессов адсорбции и десорбции. Сложность этих процессов, а также ряд неопределенностей, связанных с геометрией колонок (различия в форме и размерах зерен адсорбента, его пористости и упаковки, доступности поверхности и др.), не позволяют точно оценить влияние диффузионных и кинетических факторов и применить молекулярно-кинетическую трактовку для объяснения процессов, происходящих в хроматографической колонке. Введение коэффициента Генри имеет особый смысл, если принять во внимание динамический характер сорбционного равновесия. Последнее не следует понимать таким образом; что часть молекул сорбата все время находится в газовой фазе или растворе, а часть в сорбированном состоянии. На самом деле между сорбированными и «свободными» молекулами все время происходит обмен: одни молекулы, достигнув поверхности сорбента, поглощаются им, а другие, до этого поглощенные, пересекают поверхность раздела и переходят обратно в газовую фазу или раствор. Если следить за какой-либо молекулой, то можно будет увидеть, что часть времени tнона находится в сорбированном, а часть tп– в свободном состоянии. Чем больше tн, тем большая часть молекул находится в данное время в сорбированном состоянии, т. е. сильнее сорбция. Заполненную насадкой хроматографическую колонку промывают чистым газом-носителем. Затем, не прекращая потока газа-носителя, в колонку вводят пробу анализируемой смеси. Рассмотрим движение по колонке хроматографируемого вещества под действием потока газа-носителя, сделав следующие три, упрощающие моделирование хроматографического процесса, допущения: • хроматографическая колонка содержит неподвижную фазу и подвижную газовую фазу, которая непрерывно движется вдоль колонки со средней линейной скоростью “u ”, причем скорость потока газа-носителя остается постоянной по длине и поперечному сечению колонки в процессе всего разделения. Молекулы разделяемых соединений перемещаются вдоль колонки только в объеме газовой фазы со средней скоростью движения газа-носителя; • молекулы разделяемых соединений находятся в динамическом равновесии между газовой и неподвижной фазами, причем на состояние равновесия распределения i-компонента не оказывают влияние другие компоненты анализируемой смеси; • перепадом давления газа-носителя вдоль колонки можно пренебречь вследствие его незначительности. Температура, диаметр колонки, свойства неподвижной фазы остаются постоянными по всей длине колонки и в течение всего времени процесса разделения. С учетом отмеченных упрощений процесс перемещения хроматографируемого соединения вдоль колонки можно рассматривать как многоступенчатый процесс последовательных переходов, скачков его молекул вдоль колонки (рис. 8). 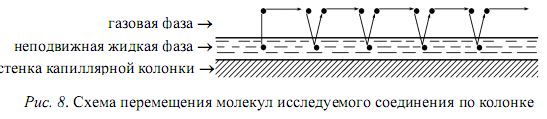 Молекулы исследуемого соединения находятся в хроматографической колонке в двух фазах: неподвижной фазе, которая сорбирует (и, следовательно, удерживает) молекулы, и подвижной газовой фазе. Отношение концентрации вещества “i ” в неподвижной фазе Сн к его концентрации в газовой фазе Сп есть величина постоянная, равная константе распределения К: Отношение числа молекул вещества, находящихся в неподвижной фазе nн, к числу молекул этого же вещества, находящихся в подвижной газовой фазе nп для данной хроматографической колонки и данных условиях разделения, есть величина постоянная и называется коэффициентом емкости данной хроматографической колонки к данному веществу в данных условиях k:  Коэффициент емкости колонки также является величиной равновесной и связан с константой распределения соотношением: 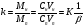 , ,где Vн и Vп объемы неподвижной и подвижной фаз в колонке; β - величина, равная отношению Vп к Vн, называемая фазовым отношением. После пребывания молекулы вещества i в адсорбированном состоянии (т.е. в неподвижной фазе) в течение некоторого среднего интервала времени н, происходит десорбция этой молекулы, которая переходит при этом из неподвижной фазы в подвижную газовую фазу. Находясь в подвижной фазе в течение некоторого среднего интервала времени п, молекула хроматографируемого вещества движется вдоль колонки в течение этого времени со средней скоростью движения потока подвижной фазы u. Затем вновь происходит сорбция молекулы неподвижной фазой и цикл сорбция – десорбция – перемещение вдоль колонки повторяется снова и снова очень большое число раз. Общую продолжительность одного цикла адсорбция–десорбция – перемещение вдоль колонки в подвижной фазе можно выразить соотношением: = п + н Теперь можно определить среднее время пребывания молекул исследуемого соединения в хроматографической колонке – t. Действительно, это время t определится как произведение общего числа скачков молекулы по всей длине колонки L на среднюю продолжительность одного элементарного цикла: 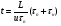 . . С учетом выражения для коэффициента емкости колонки k' = nн/ nп = н/ п , (12 ) получаем основное уравнение для вычисления времени нахождения исследуемого соединения в хроматографической колонке: 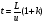 . (13) . (13)Численное значение 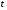 определяется из экспериментально полученной выходной кривой. определяется из экспериментально полученной выходной кривой.Названные параметры удерживания являются абсолютными параметрами удерживания, поскольку они характеризуют не только интенсивность взаимодействия разделяемых веществ с неподвижной фазой, но и процессы, учитывающие пребывание разделяемых веществ в газе-носителе во внутреннем объеме хроматографической колонки. Абсолютные значения параметров удерживания существенно зависят от большого числа параметров, характеризующих условия процесса разделения, и поэтому не могут выступать в качестве индивидуальных характеристик разделяемых компонентов. Поскольку разделяемые соединения не вступают в какие-либо ощутимые взаимодействия с газом-носителем, время пребывания каждого из них в объеме газа-носителя в колонке является постоянным, одинаковым для всех разделяемых компонентов и определяется величиной внутреннего объема колонки, не заполненного твердой насадкой и скоростью потока подвижной фазы. Время пребывания разделяемых компонентов в неподвижной фазе, напротив, зависит только от индивидуальных особенностей взаимодействия разделяемых компонентов с неподвижной фазой. Вследствие различия этих особенностей взаимодействия для разделяемых веществ оно оказывается различным и является индивидуальной характеристикой разделяемых соединений на данной неподвижной фазе в данных условиях, используемой для качественного анализа.  Откуда величина коэффициента емкости колонки по отношению к исследуемому соединению выразится соотношением: 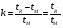 . .  где t − время удерживания, равное интервалу времени от момента ввода пробы в колонку до момента выхода из нее максимума пика хроматографируемого вещества “i ”; tо − время удерживания несорбирующегося компонента; t′ − исправленное, или приведенное, время удерживания, равное интервалу времени от момента выхода максимума пика несорбирующегося компонента до выхода максимума пика хроматографируемого вещества i. Анализ уравнений указывает на зависимость времени удерживания одного и того же соединения от условий процесса разделения, которые определяют время удерживания несорбирующегося компонента, величину фазового отношения и, что самое важное, влияние различия в величинах констант распределения разделяемых соединений между фазами на эффективность их разделения. Таким образом, поскольку компоненты анализируемой смеси в колонке образуют с неподвижной фазой различные по силе связи, то вследствие движения газа-носителя постепенно перемещаются вдоль слоя насадки с различными для каждого компонента скоростями. В результате, зона вещества, образующего более прочные связи с неподвижной фазой, постоянно отстает от зоны вещества, образующего менее прочные связи, и при достаточной длине хроматографической колонки смесь веществ разделяется. Время удерживания несорбирующегося компонента может быть определено или экспериментально, или расчетным методом. При экспериментальном определении времени удерживания несорбирующегося компонента в хроматографическую колонку вводятся вещества с минимальной молярной массой, способные регистрироваться используемым детектором. Так, при использовании детектора по теплопроводности в качестве несорбирующегося компонента используется воздух, а при использовании пламенно-ионизационного детектора метан. Время выхода несорбирующегося компонента зависит от плотности упаковки насадки в колонке и длины колонки: для колонок одинаковой длины с ростом плотности упаковки насадки в колонке это время будет уменьшаться. Рассмотрим природу сил, заставляющих молекулы концентрироваться на поверхности адсорбента или поглощаться пленкой неподвижной фазы. В общем силы взаимодействия между сорбатом и сорбентом можно назвать физическими или ван-дер-ваальсовыми. Это те же силы, которые проявляются в жидкостях, удерживая молекулы последних друг возле друга, в сжатых газах, вызывая отклонение их поведения от законов идеальных газов, в некоторых так называемых молекулярных кристаллах, например твердой углекислоте. |