хроматография-лекции. Хроматографические методы. Общая характеристика методов
 Скачать 6.82 Mb. Скачать 6.82 Mb.
|

Метод простой нормировки можно использовать при количественном обсчете хроматограмм в том случае, когда есть уверенность в том, что все вещества анализируемой пробы, взятые в равном количестве, дают на хроматограмме пики, имеющие равные площади, т. е. что чувствительность детектора хроматографа ко всем компонентам анализируемой пробы одинакова. Это условие достаточно строго выполняется, если вещества сходны по химическому строению. Площади пиков рассчитывают, умножая высоту пика на ширину, измеренную на его полувысоте. Процентное содержание данного компонента в анализируемой смеси находят по формуле: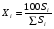 (1.69) (1.69)где Xi процентное содержание i-ого компонента; Si площадь пика определяемого компонента; Si сумма площадей пиков всех компонентов. Метод простой нормировки не дает точных результатов в случае различной чувствительности детектора по отношению к различным компонентам смеси. В этом случае следует применять метод внутренней нормировки с калибровочными коэффициентами. Этот метод учитывает различия чувствительности детектора по отношению к компонентам анализируемой смеси. Он более точен и надежен, чем метод простой нормировки. Количественный обсчет хроматограмм ведут по формуле 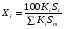 , (1.70) , (1.70)где Кi калибровочный коэффициент, мг/см2. Если отдельные компоненты разделяются не полностью, то пользуются следующей формулой: 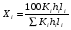 , (1.71) , (1.71)где hi высота пика i-ого компонента; li расстояние удерживания этого компонента (по диаграммной ленте). Физический смысл калибровочного коэффициента Ki количество вещества, соответствующее единице площади пика. Практически калибровочный коэффициент рассчитывают следующим образом. Приготовив искусственную смесь определенного состава, снимают хроматограмму. Разделив массу каждого компонента в пробе этой смеси на площадь соответствующего пика компонента, получают калибровочный коэффициент Ki (мг/см2). Для определения Ki часто используют так называемые поправочные коэффициенты Ki’, т.е. калибровочные коэффициенты Ki хроматографируемых веществ, к калибровочному коэффициенту Kст. вещества, выбранного в качестве стандарта: 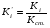 . (1.72) . (1.72)K’ст является безразмерной величиной. Применение метода внутренней нормировки при использовании большинства детекторов часто не требует специальной калибровки, так как калибровочные коэффициенты для многих веществ даны в литературе. Для быстрого определения приведенной площади пика КiSi можно воспользоваться имеющимися в литературе номограммами. Достоинство метода внутренней нормировки состоит в том, что искажения, имеющиеся в одинаковой степени у всех пиков, в конечном счете не влияют на точность результатов. Ошибки этого метода могут быть связаны с постепенным изменением режима в процессе анализа. 7. Практическое использование хроматографии в контроле качества продукции В настоящее время разработаны хроматографические методики, предназначенные для определения состава многих видов пищевой и промышленной продукции или установления содержания в них отдельных компонентов. Многие из этих методик гостированы и их применение регламентировано в качестве метода контроля показателей качества соответствующей продукции. Например, в соответствии с ГОСТ 30536-97 контроль качества этилового спирта, должен проводиться газовой хроматографией на колонке с неподвижной жидкой фазой ПЭГ-2000. Качество авиационных и автомобильных бензинов, согласно с СТБ 1276-2001, должно контролироваться газохроматографически на капилярных колонках длиной 50 метров с неподвижной жидкой фазой полидиметилсилоксаном. Эти методики позволяют количественно определить состав контролируемых продуктов по основным компонентам и установить содержание в них многих примесей, оказывающих негативное влияние на качество продукции. Однако большинство хроматографических методик предназначено для определения в многокомпонентной пищевой или промышленной продукции содержания отдельных компонентов. К таким методикам относятся, например, методики определения в пищевых продуктах остаточных пестицидов, афлатоксинов, витаминов, отдельных жирных кислот, отдельных сахаров, насыщенных и ароматических углеводородов и многих других. Их особенностью является то, что ввиду многокомпонентности и сложности состава анализируемых продуктов на стадии подготовки пробы к анализу тем или иным способом, чаще всего жидкостной экстракцией, проводится выделение определяемого компонента, чистого или содержащего небольшое количество примесей, из анализируемого продукта, а затем на хроматографической колонке осуществляется его качественная идентификация и количественное определение. Во многих случаях для выделения определяемых компонентов и оптимизации условий хроматографирования приходится подвергать определяемые компоненты дополнительной обработке, позволяющей, например, отделить их от компонентов, мешающих определению или засоряющих хроматографическую колонку, или придать определяемым компонентам необходимые для хроматографирования свойства, например, более высокую летучесть. Например, при хроматографическом определении насыщенных и ароматических углеводородов в продуктах животного и растительного происхождения, рыбопродуктах, определяемые компоненты выделяют из неомыляемой фракции липидов, получаемой при обработке анализируемого образца спиртовым раствором щелочи, экстракцией гексаном. Затем экстракт разделяют на хроматографической колонке и углеводороды идентифицируют и количественно определяют с помощью газовой или высокоэффективной жидкостной хроматографии. Для газохроматографического определения сахаров глюкозы, фруктозы, арабинозы, ксилозы, галактозы, сахарозы, мальтозы, лактозы, рафинозы, сорбита и инозита в пищевых продуктах для повышения летучести сахаров анализируемые пробы обрабатывают триметилхлорсиланом или гексаметилдисилазаном в присутствии пиридина и трифторуксусной кислоты. Затем 1 мкл пиридинового раствора триметилсилильных производных углеводов вводят при температуре 250 оС в испаритель хроматографа и элюируют газом-носителем, подаваемым со скоростью 40 см3/мин через колонку длиной 2 м с внутренним диаметром 3 мм при программированном подъеме температуры в колонке со скоростью 4оС/мин от 125 до 270оС. Идентификацию индивидуальных триметилсилильных производных углеводов проводят по времени удерживания триметилсилильных производных сахаров-метчиков. При газохроматографическом определении состава жирных кислот в продуктах питания липиды выделяют экстракционным методом, наиболее подходящим для конкретной группы продуктов и исключающим термическое воздействие на объект. Затем проводят омыление образцов, в результате которого происходит расщепление триглицеридов и образование свободных жирных кислот. Заключительной стадией пробоподготовки является метилирование (получение метиловых эфиров) жирных кислот, которое проводится для придания большей летучести определяемым компонентам и, соответственно, для снижения температуры хроматографической колонки, что необходимо для предотвращения химических превращений определяемых кислот в жестких температурных условиях. Разделение смеси метиловых эфиров жирных кислот осуществляют в насадочной колонке из стекла или нержавеющей стали длиной 13 м с внутренним диаметром 24 мм или в капиллярной колонке из стекла, кварца, меди или нержавеющей стали длиной 2050 м с внутренним диаметром 0,30,5 мм при температуре 2201оС. В насадочной колонке в качестве неподвижной твердой фазы используют нейтральный носитель, а в качестве неподвижной жидкой фазы полиэтиленгликольадипат, полиэтиленгликольсебацинат, другие полиэфиры, цианосиликоны, силары или другие жидкости. Количество неподвижной жидкой фазы должно составлять 520% от массы инертного носителя. Толщина пленки неподвижной жидкой фазы в капиллярных колонках должна быть 0,11,5 мкм. Детектирование проводится пламенно-ионизационным детектором при температуре детектора 230235оС. Необходимо отметить, что применение хроматографии для контроля качества продукции достаточно быстро расширяется по мере оснащения производственных и испытательных лабораторий современным оборудованием. |