хроматография-лекции. Хроматографические методы. Общая характеристика методов
 Скачать 6.82 Mb. Скачать 6.82 Mb.
|

Часть сорбированных примесей удаляется с поверхности при промывке раствором ведущего электролита (если сорбция обратимая), и, подбирая время промывки, удается при последовательных анализах сохранять постоянными времена миграции компонентов. Если же в пробах имеются примеси, сорбирующиеся практически необратимо, приходится периодически промывать капилляр растворами, которые способны удалить накопившиеся примеси, и далее снова кондиционировать капилляр относительно раствора ведущего электролита. Эффективным средством борьбы с такими примесями является их предварительное удаление на этапе подготовки пробы к анализу. |
| Детектирование | Селек- тив ность | Универ-саль-ность | Качест-венная инфор-мация о веществе | Детек-тирова-ние в капил-ляре | Примерный предел детектирования, моль/л | Частота исполь-зования, % |
| Прямое фотометрическое в УФ-области *(для диодной матрицы) | + | — | + * | + | 10-5-10-7 | 55 |
| Косвенное фотометри-ческое в УФ-области | — | + | — | + | 10-4-10-6 | 5 |
| Флуориметрическое: прямое косвенное | + — | — + | — — | + + | 10-7-10-9 10-6-10-8 | 15 2 |
| Индуцированное лазером флуориметрическое | + | — | — | + | 10-13-10-16 | 5-7 |
| Масс-спектрометрическое | + | + | + | — | 10-8-10-10 | 10 |
| Амперометрическое: прямое косвенное | + — | — + | — — | — — | 10-7-10-10 10-6-10-8 | 2 <1 |
В капиллярном электрофорезе используют те же принципы детектирования, что и в ВЭЖХ. Важным преимуществом КЭ перед ВЭЖХ, помимо плоского профиля ЭОП, является отсутствие соединительных гидравлических линий между узлами ввод пробы–капилляри капилляр–детектор, которые в случае ВЭЖХ могут приводить к уширению зоны вещества за счет внеколоночного размывания.
Основным вариантом детектирования является фотометрическое, основанное на поглощении веществом УФ или видимого света. Фотометрические детекторы в КЭ разделяются на несколько типов.
Детекторы с фиксированной длиной волны:источники света с линейчатым спектром ртутная лампа (254 нм), кадмиевая лампа (229 нм) и цинковая лампа (214 нм). Это наиболее простые системы; в приборах «Капель-103, -104» фотометрический детектор работает при = 254 нм, поэтому отклик детектора будет наблюдаться только тогда, когда определяемый компонент имеет заметное поглощение для указанной . Этот случай называется прямым детектированием, электрофореграмма представляет собой набор положительных пиков, возвышающихся над базовой линией. Так определяются органические соединения с ароматической структурой или сопряженными двойными связями, некоторые неорганические соединения и др.
Детекторы с изменяемой длиной волны:источниками света служат дейтериевые (190–350 нм) и вольфрамовые (340–850 нм) лампы. Необходимая спектральная селекция достигается применением монохроматоров или узкополосных светофильтров.
В детекторах на диодной матрице (ДДМ) световой поток, прошедший через капилляр, разлагается в спектр с помощью высококачественного светосильного монохроматора, а матрица фотодиодов регистрирует сигналы в УФ и видимой частях спектра (УФ-В-детекторы), обеспечивая запись в режиме сканирования. Данные, полученные одновременно на 25 различных , обрабатываются компьютером, выделяющим сигнал при оптимальной и вычитающим фон. Применение ДДМ обеспечивает получение аналитических данных высокой достоверности. Например, при определении гомогенности (однородности) пика осуществляется спектральный контроль в максимуме и по обоим склонам пика. Если пик однороден, то все три спектра идентичны. Для индивидуального вещества отношение высот пиков на электрофореграммах, записанных при двух различных , есть величина постоянная. Гомогенность пика проверяется также при сравнении параметров миграции соединения, полученных при двух разных (для ДДМ обе электрофореграммы получаются в ходе одного анализа). Идентификацию пика проводят путем сравнения времен миграции и спектров стандарта и компонента пробы.
Для соединений, анализируемых с помощью КЭ и не поглощающих в УФ-диапазоне, возможна регистрация методом косвенного УФ-детектирования. В этом случае в состав ведущего электролита вводят хромофор вещество, поглощающее на требуемой . Так, в случае определения анионов используют поглощающий анион, например, CrO42 или фталат, а при определении катионов используют катионы ароматических аминов или гетероциклов, в частности, катион бензимидазолия. Так как ионная сила ведущего электролита в процессе разделения остается постоянной, в зоне, где находится непоглощающий ион, уменьшается концентрация поглощающего иона. Обмен происходит строго эквивалентно, на электрофореграмме наблюдаются обратные (отрицательные) пики, площади которых пропорциональны концентрациям ионов. Косвенное УФ-детектирование является универсальным и позволяет регистрировать все присутствующие в анализируемом растворе ионы.
Системы термостабилизации. Сбор и обработка данных
Наиболее простым вариантом охлаждения капилляра является интенсивный обдув комнатным воздухом. Дополнительно используют жидкостные системы термостатирования с диапазоном температур 470°С 0,1°С. Как правило, термостатируют только капилляры. Термостатирование автозагрузчика пробы ценно при анализе термолабильных образцов. После загрузки в автосемплеры проб, рабочих буферов и вспомогательных растворов измерения проводятся в автоматическом режиме по заданным параметрам.
Все приборы КЭ комплектуются программными продуктами, позволяющими записывать данные, проводить их качественную и количественную обработку, формировать отчеты. Некоторые программы способны также управлять системами капиллярного электрофореза.
Эффективность разделения
Метод КЭ характеризуется высокой эффективностью. Эффективность N, выраженная числом теоретических тарелок, может быть определена непосредственно из электрофореграммы по уравнению (1):
N = 5,55 * (tм / W1/2)2 (1)
где tм время миграции аналита, W1/2 ширина пика на ½ высоты.
Основными причинами, приводящими к снижению N, являются:
величина зоны вводимой пробы, определяемая длительностью ввода: в идеале она должна быть как можно меньше, однако достижение низких пределов обнаружения требует увеличения объема пробы или ее концентрирования;
температурный градиент: при анализе в капилляре протекает электрический ток, величина которого зависит от удельной проводимости буфера и диаметра капилляра (D). Отвод тепла происходит через стенки капилляра, что приводит к возникновению в буфере радиального температурного градиента. Разница в температуре между серединой и стенками капилляра возрастает пропорционально D2. Температура в центре капилляра может быть на 10°С выше, чем на внутренней стенке. Возникающий вследствие этого градиент вязкости приводит к тому, что вещество у стенки перемещается медленнее, чем в центре, что влечет за собой уширение полос и снижение эффективности;
адсорбция на стенках капилляра взаимодействие веществ со стенками капилляра ведет к искажению формы пиков («хвосты»);
различия в электропроводностипробы и ведущего электролита: уширение пика, обусловленное электрофоретическими эффектами, пропорционально проводимости раствора образца относительно буфера. В случае высокой концентрации пробы градиент потенциала (и линейные скорости ионов) в зоне образца заметно ниже, чем в ведущем электролите. Благодаря этому происходит дестэкинг уширение пиков. Обратная ситуация в соотношении проводимостей (стэкинг), наоборот, приводит к формированию узких пиков на электрофореграмме;
различия в уровне буферов во входном и выходном сосудах приводят к возникновению гидродинамического потока с параболическим профилем; чем больше диаметр капилляра, тем значительней это сказывается на эффективности разделения.
продольная диффузия в КЭ практически не дает уширения зоны вещества, что в основном обусловлено плоским профилем ЭОП.
Чувствительность метода
Основным способом детектирования в КЭ является фотометрический, чувствительность которого не всегда достаточна, поскольку детектирование происходит в слое малого внутреннего диаметра капилляра с низкой концентрацией пробы.
Подходы к увеличению чувствительности делятся на 3 категории: стратегия концентрирования образца; увеличение длины оптического пути; использование высокочувствительных селективных детекторов.
Стэкинг (stacking) один из наиболее общих подходов к увеличению концентрационной чувствительности в КЭ. Стэкинг образца происходит, когда ионы аналитов пересекают границу, которая отделяет зону низкой проводимости раствора образца и высокой ведущего электролита. В случае если матрица образца имеет значительно более низкую проводимость, чем ведущий электролит, в зоне образца возникает относительно высокое электрическое поле. Аналиты внутри зоны образца движутся с более высокой локальной скоростью, и, замедляясь на границе с зоной ведущего электролита, концентрируются. Стэкинг образца применителен только к заряженным аналитам.
Свипинг (sweeping) техника концентрирования нейтральных частиц в МЭКХ, суть которой заключается в том, что аналиты концентрируются псевдостационарной фазой (мицеллой), которая проникает в зону образца, где мицеллы отсутствуют. При этом (в отличие от стэкинга) проводимость раствора образца близка проводимости ведущего электролита. В ряде случаев свипинг позволяет получать 100-кратные концентрирования (on-line).
Чувствительность метода КЭ с УФ-детектированием может быть повышена за счет увеличения длины оптического пути: зону детектирования выполняют в форме пузырька (увеличение сигнала в 3–5 раз), используют капилляры Z-формы (увеличение сигнала в 20–40 раз).
Чувствительности определения способствует снижение уровня шума детектора за счет стабилизации светового потока ламп и учета флуктуаций интенсивности потоков в каналах фотометра.
Разрешение и селективность разделения
Полное или частичное разделение компонентов пробы характеризуется параметром Rs (разрешение). Чаще всего в КЭ разрешение двух компонентов определяют так же, как в ВЭЖХ:
Rs = 2 * (t1 – t2) / (W1 + W2) (2)
где t1 и t2 времена миграции компонентов, мин.;
W1 и W2 — ширина пиков 1 и 2 при основании, мин.
Разрешение в КЭ, в основном, управляет эффективностью, а не селективностью. В этом заключается отличие КЭ от ВЭЖХ, где картина прямо противоположная. Благодаря узким зонам компонентов в КЭ даже очень малые различия в электрофоретической подвижности веществ (<0,05 %) оказываются достаточны для полного разделения.
Если выразить Rs через эффективность:
Rs = 0,25 * (N)1/2 * ( / ср) (3)
где = 1 2, а ср = (1 + 2) / 2
то становится очевидным, что в противоположность эффективности, линейно возрастающей при увеличении рабочего напряжения, разрешение будет расти не так заметно.
Существующее многообразие вариантов КЭ обеспечивает различную селективность разделения вследствие отличающихся механизмов разделения. Так, в зонном варианте КЭ при разделении компонентов 1 и 2 фактор селективности (α) определяется выражением:
α = 1 / 2 (4)
где 1 и 2 электрофоретические подвижности компонентов 1 и 2.
При концентрации ПАВ в растворе электролита больше ККМ реализуется вариант МЭКХ с главным принципом разделения на основе распределения компонентов пробы между гидрофильной (водной) и гидрофобной (мицеллярной) фазами, селективность которого будет определяться отношением факторов емкости двух компонентов: α = k'2 / k'1. В режиме МЭКХ k' можно найти по уравнению (5):
k' = (ta – t0) / t0 * (1 – ta / tm) (5)
где ta время миграции анализируемого вещества,
t0 tудерж компонента, абсолютно не удерживаемого мицеллой,
tm tудерж компонента, полностью удерживаемого мицеллой.
Для нахождения t0 и tm в пробу вводят маркер ЭОП (ацетон) и метку мицелл (судан 3 или судан 4), соответственно.
Задача повышения селективности разделения в КЭ требует знания факторов, ее определяющих, и может быть решена за счет изменения рН ведущего электролита, введения в состав буфера различных добавок ПАВ, макроциклов, органических растворителей (табл. 3). Скорость ЭОП не изменяет селективности разделения и определяет лишь изменение времени миграции на равную величину для всех компонентов пробы.
Таблица 3.
Факторы, определяющие параметры разделения в методе КЭ.
| Факторы селективности | Характер влияния |
| рН ведущего электролита | Изменение заряда и формы нахождения вещества в растворе, скорости ЭОП |
| Ионная сила ведущего электролита | Изменение скорости ЭОП |
| Напряжение | Скорость ЭОП и электромиграции ионов, температурный градиент в капилляре |
| Параметры гидродинамического или электрокинетического способа ввода пробы | Селективное концентрирование при вводе пробы, объемная и концентрационная перегрузка системы разделения |
| Длина и внутренний диаметр капилляра | Изменение времени анализа и скорости ЭОП, температурный градиент |
| Температура | Вязкость электролита, сольватация и химические равновесия |
| Добавка ПАВ в ведущий электролит | Мицеллообразование при ККМ. Ион-парные взаимодействия. Обращение ЭОП |
| Добавка органических растворителей в ведущий электролит | Преимущественно снижение скорости ЭОП. Сольватация |
| Добавка макроциклов в ведущий электролит | Образование комплексов включения. Растворение гидрофобных компонентов Селективное хиральное распознавание |
Ведущий электролит чрезвычайно важен для успешного разделения в любом варианте КЭ. Величина рН электролита определяет как скорость течения жидкости в капилляре (величину ЭОП), так и заряд компонента в растворе. Чувствительность ЭОП к изменению рН раствора предполагает использование ведущих электролитов с высокой буферной емкостью и диапазоном рН равным рКа ± 1. Вследствии высокой стабильности кварцевого капилляра, при электрофоретическом разделении можно использовать буферные системы с рН от 2 до 12. Среди наиболее распространенных электролитов следует упомянуть фосфатный (рК1 = 2,1 и рК2 = 7,2), ацетатный (рКа = 4,7), имидазольный (рКВН+ = 7,0), триоксиметиламинометановый (TRIS, рКВН+ = 8,1), боратный (рК1 = 9,2) и 2-(N-циклогексиламино)этансульфонатный (CHES, рКа = 9,5) буферные растворы.
Буфер для проведения КЭ должен обладать достаточной буферной емкостью в выбранном диапазоне рН, иметь малое поглощение на длине волны детектирования и низкую подвижность ведущего иона.
Список оптимальных буферов возглавляют боратный буфер и TRIS, так как они могут использоваться в широком диапазоне концентраций без существенного увеличения тока, что позволяет, в свою очередь, применять максимально высокие напряжения в ходе анализа.
Среди используемых в КЭ добавок наиболее популярны ПАВ. Их введение в состав буферов позволяет влиять на селективность разделения, причем определяющими факторами являются тип и концентрация ПАВ. В КЭ используются ионогенные (катионные КПАВ и анионные АПАВ), а также цвиттер-ионные и нейтральные ПАВ.
При концентрации ниже ККМ мономерные формы ионогенных ПАВ модифицируют стенки капилляра (ЦТАБ), влияют на поведение зоны пробы в капилляре и на стенки самого капилляра, модифицируя ЭОП (уменьшая, увеличивая или обращая).
Органические растворители (метанол, ацетонитрил и др.), которые вводят в буферный раствор в концентрации до 30 %, повышают растворимость анализируемых соединений, делая КЭ пригодным для анализа веществ с ограниченной растворимостью в водных средах.
Макроциклические реагенты как компоненты ведущих электролитов широко распространены в КЭ. Макроциклическими называют органические соединения, молекулы которых содержат не менее 9 атомов в цикле, причем не менее 3 из них гетероатомы (О, N и S). К макроциклическим соединениям относят циклодекстрины, краун-эфиры, криптанды, каликсарены и др. Все они способны взаимодействовать как с неорганическими веществами, так и с органическими субстратами различной природы, при этом происходит включение фрагментов анализируемых веществ в полость макроцикла (МЦ). В образующихся при этом комплексах включения по типу «гость–хозяин» «хозяином» служит макроцикл, а «гостем» субстрат. В зависимости от своего строения МЦ могут связывать молекулярные, катионные или анионные субстраты за счет нековалентных взаимодействий: ион-ионных, ион-дипольных, гидрофобных, водородных связей.
Использование макроциклических добавок для хиральных и ахиральных разделений возможно в зонном и мицеллярном вариантах, причем селективность последнего будет выше за счет распределения компонентов смеси между тремя фазами: водной, псевдостационарной мицеллярной и псевдостационарной фазой макроцикла.
Обработка результатов в капиллярном электрофорезе.
Качественный и количественный анализ
Для качественного и количественного анализа в КЭ обязательно проводят градуировку системы путем анализа нескольких смесей известного состава. Результатом градуировки являются формирование таблицы компонентов, их времен миграции и построение градуировочной зависимостисигнала детектора от концентрации вещества.
В КЭ используют те же принципы интегрирования пиков, методы градуировки, способы формирования отчетов, как в газовой хроматографии и ВЭЖХ. По аналогии с ВЭЖХ большинство детекторов в КЭ являются концентрационными, где высота или площадь пика прямо пропорциональны концентрации вещества, образующего пик.
Качественный анализ обычно состоит в сравнении времен миграции (для КЗЭ) или времен удерживания (для МЭКХ, из-за различий в принципе разделения), полученных для стандарта и пробы, измеренных в одинаковых условиях. При совпадении этих времен с заданной точностью (окно идентификации редко >5 %), то считают, что искомое вещество в пробе найдено. Такой способ идентификации не надежен в случае анализа проб со сложной матрицей.
При качественном анализе близких пиков рекомендуется использование метода добавок. Если на электрофореграмме появляется новый пик, это означает, что анализируемый компонент ранее в пробе отсутствовал. Если же один из бывших пиков увеличился по высоте (площади), то можно утверждать, что это и есть анализируемый компонент. Величину добавки обычно выбирают так, чтобы высота (площадь) интересующего нас пика увеличилась не более чем в 2–3 раза.
Ситуация, когда время миграции компонента нестабильно от анализа к анализу, нередко связана с нестабильностью ЭОП. Использование в этих случаях маркера ЭОП (например, ацетона) как в растворе стандарта, так и в пробе, позволяет вычислить исправленные времена миграции, представляющие собой разность времен миграции анализируемого вещества и метки ЭОП.
Для повышения достоверности идентификации компонента используют введение в стандартный раствор и пробу внутреннего стандарта (маркера). Это вещество, заведомо отсутствующее в пробах, но имеющее схожие с определяемым компонентом аналитические свойства. Для стандарта и пробы вычисляют относительные времена миграции и находят в пробе близкие по значению результаты.
Наиболее достоверную идентификацию вещества можно получить при использовании диодно-матричного детектора, который по результату одного анализа может предоставить информацию:
- по сопоставлению времени миграции вещества и его спектра в пробе и стандартном растворе (при этом дополнительно будет дана оценка чистоты пика пробы по наложению спектров, снятых в трех точках пика: на обоих склонах и в максимуме);
- по отношению площади пика на двух разных , полученных для стандарта и пробы. Для одного и того же вещества на двух разных при неизменном времени миграцииотношение площадей в стандартном растворе и растворе пробы должно быть постоянным. Длины волн выбирают так, чтобы компонент имел при них разное поглощение и высота или площадь пика при разных были бы различными.
Известно также, что площадь пика зависит от ЭОП и электрофоретической подвижности иона, которые влияют на его скорость. Чем медленнее движется ион по капилляру, тем шире пик и больше его площадь. Для корректировки нестабильности скорости движения иона рекомендуется сравнивать для двух разных отношения площади пика к его времени миграции. Считается также, что использование электрофоретической подвижности вместо времени миграции позволяет корректно идентифицировать компоненты сложных смесей.
Количественная обработка результатов анализа
Для количественного определения необходимо выбрать метод градуировки (внешнего стандарта (абсолютной градуировки), внутреннего стандарта, метод добавок) и определить, какую величину отклика детектора высоту пика или площадь пика будут использовать. Затем анализируют стандартные растворы с известными концентрациями веществ и для каждого компонента строят градуировочную зависимость отклика детектора от концентрации вещества, после чего анализируют пробу неизвестного состава и по градуировочному графику находят концентрацию определяемых веществ.
Основным методом градуировки является метод внешнего стандарта(абсолютной градуировки), для которого необходимо иметь ГСО или химически чистые стандарты всех определяемых компонентов. Для одноточечной градуировки компонента используется один градуировочный раствор, зависимость носит строго линейный характер и, как правило, выходит из начала координат. Для построения многоточечной градуировки анализируют несколько подобранных по концентрациям градуировочных растворов, после чего с помощью метода наименьших квадратов рассчитывают коэффициенты прямой, наилучшим образом описывающей экспериментальные данные.
Современные программные комплексы позволяют собирать и обрабатывать электрофоретические данные, хранить их, а также формировать и выдавать отчеты. Для систем КЭ «Капель» рекомендуется программа «МультиХром®». Приборы «Капель» модификации «М» снабжаются программой «Эльфоран®», которая позволяет также управлять самой системой капиллярного электрофореза.
Объекты для анализа методом КЭ. Подготовка пробы
Первые аналитические приложения КЭ были связаны с разделением заряженных компонентов: наиболее подходящими оказались неорганические катионы и анионы, а также карбоновые кислоты. В биотехнологии КЭ используют для анализа макромолекул: белков, углеводов, нуклеиновых кислот. В фармации оценка чистоты лекарственных препаратов и хиральные разделения до сегодняшнего дня в мире на 90 % выполняются различными вариантами КЭ.
Первым этапом анализа является отбор и подготовка пробы. Отобранная проба должна быть представительной, а процедура отбора пробы легко воспроизводимой.
Схема анализа пробы, разбавленной буфером с минимальной электропроводностью при известном ионном составе включает этапы:
а) для анализа катионов используют 25мМ фосфатный буфер (рН 2,5);
б) для анализа анионов используют 25мМ боратный буфер (рН 9,3);
в) для анализа нейтральных соединений используют 25мМ боратный буфер (рН 9,3) с 25мМ додецилсульфата натрия.
При наложении пиков проводят оптимизацию разделения.
При отсутствии пиков из-за слишком низкой концентрации компонентов выбирают более чувствительный детектор, концентрируют пробу или используют электрокинетический ввод пробы.
На этапе подготовки пробы к анализу проводят удаление мешающих веществ, выделение и концентрирование определяемых соединений, их превращение в более удобные аналитические формы (при необходимости). Так, КЗЭ с косвенным УФ-детектированием позволяет анализировать неорганические анионы в водных объектах на уровне 0,1 мг/л. Подготовка образца питьевой, природной или сточной воды к анализу заключается в фильтровании пробы через мембранный фильтр (диаметр пор 0,2 мкм) и дегазировании фильтрата путем центрифугирования. Для КЭ характерны высокая скорость анализа и малый расход реактивов. Промежуточные операции пробоподготовки должны быть унифицированными и максимально простыми.
Области применения метода капиллярного
электрофореза и примеры использования
Анализ объектов окружающей среды.
Анализ NH4+, щелочных и щелочноземельных металлов (рис. 9-10)
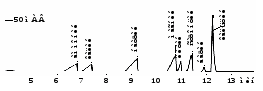
Рис. 9. Электрофореграмма модельного раствора катионов.
«Капель-105», капилляр: внутр. диаметр 75 мкм, Lэфф / Lобщ = 50/60 см.
Ведущий электролит: 6 мМ БИА, 2,5 мМ винная кислота, 2 мМ 18-краун-6.
Ввод пробы: гидродинамический 150 мбар*с. Напряжение: +10 кВ.
Температура: 20°С. Детектирование: 254 нм, косвенное.
Определение 8 катионов в одной порции пробы. Макроцикл 18-краун-6 используется как селективная добавка для разделения К+ и NH4+. Методики определения катионов щелочных и щелочноземельных металлов и важнейших неорганических анионов методом КЭ нашли широкое применение в экологическом контроле природных, питьевых, сточных и технологических вод, метрологически аттестованы и реализованы на системах КЭ «Капель».
Анализ NH4+, K+, Na+, Mg2+, Ca2+в реальных водах
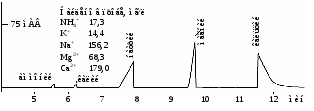
Рис. 9. Электрофореграмма очищенной сточной воды (разбавлена в 5 раз).
«Капель-105», капилляр: внутр. диаметр 75 мкм, Lэфф / Lобщ = 50/60 см.
Ведущий электролит: 10 мМ БИА, 5 мМ винная кислота, 2 мМ 18-краун-6.
Ввод пробы: гидродинамический 300 мбар*с. Напряжение: +13 кВ.
Температура: 20°С. Детектирование: 254 нм, косвенное.
Предложенная схема анализа применима к питьевым, природным и сточным водам. Подготовка пробы заключается в обязательном ее фильтровании, при необходимости разбавлении, центрифугировании.
Анализ неорганических анионов с обращением ЭОП (рис. 9)
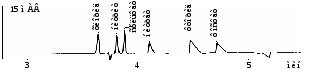
Рис. 9. Электрофореграмма модельного раствора анионов, модификатор электроосмотического потока ЦТАБ.
«Капель-105», капилляр: внутр. диаметр 75 мкм, Lэфф / Lобщ = 50/60 см.
Ведущий электролит: 5 мМ СrO3, 20 мМ ДЭА, 1,7 мМ ЦТАБ.
Ввод пробы: гидродинамический 300 мбар*с. Напряжение: -18 кВ.
Температура: 20°С. Детектирование: 254 нм, косвенное.
При анализе анионов применяют косвенное детектирование. Модификатор ЭОП цетилтриметиламмония бромид (ЦТАБ) дает отрицательный пик Br между пиками Cl и NO2. Последний отрицательный пик обусловлен избытком HCO3 в буфере по сравнению с пробой.
Анализ неорганических анионов без обращения ЭОП (рис. 9)
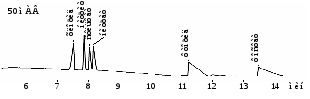
Рис. 9. Электрофореграмма раствора анионов без модификатора ЭОП.
«Капель-105», капилляр: внутр. диаметр 75 мкм, Lэфф / Lобщ = 50/60 см.
Ведущий электролит: 5 мМ СrO3, 20 мМ ДЭА. Ввод пробы: гидродинамический 300 мбар*с. Напряжение: -18 кВ. Температура: 20°С.
Детектирование: 254 нм, косвенное.
Разделение неорганических анионов можно проводить без модификации внутренней поверхности капилляра, с отрицательной полярностью напряжения и косвенным режимом детектирования.
Анализ неорганических катионов в яблочном соке (рис. 9)
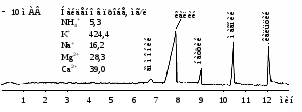
Рис. 9. Электрофореграмма неорганических катионов в яблочном соке.
«Капель-105», капилляр: внутр. диаметр 75 мкм, Lэфф / Lобщ = 50/60 см.
Ведущий электролит: 6 мМ БИА, 2,5 мМ винная кислота, 2 мМ 18-краун-6. Ввод пробы: гидродинамический 200 мбар*с. Напряжение: +12 кВ. Температура: 20°С. Детектирование: 254 нм, косвенное.
Определение неорганических катионов в свежевыжатых и консервированных соках (разбавление 1:9).
Анализ ионного состава воды. Определение неорганических
катионов (NH4+, K+, Na+, Li+, Mg2+, Sr2+, Ba2+, Ca2+).
Особенности методики, практические рекомендации
Здесь будет рассмотрен вариант одновременного определения ряда катионов и анионов с использованием прибора «Капель-103РЕ».
Для определения катионов щелочных и щелочноземельных металлов используют источник высокого напряжения положительной полярности. Это классическая схема (Рис. 7), где детектор находится вблизи катода и ЭОП движется от анода к катоду. В этом случае катионы движутся к катоду в направлении ЭОП, но быстрее него.
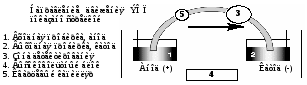
Рис. 7. Классическая схема прибора для анализа неорганических
катионов (с положительным высоковольтным блоком).
Для регистрации пиков катионов применяют косвенное детектирование: в состав ведущего электролита вводят поглощающий катион бензимидазола (БИА) в концентрации 0,01 М, обеспечивающей необходимую оптическую плотность исходного раствора. При разделении катионы пробы эквивалентно замещают в растворе катион бензимидазолия, что приводит к снижению оптической плотности в зоне каждого катионного компонента.
При косвенном детектировании электрофореграмма представляет собой базовую линию с отрицательными пиками. Программы обработки данных имеют опцию «Перевернуть» и позволяют представить электрофореграмму в привычном виде с «положительными» пиками.
Бензимидазол в водном растворе является слабым основанием с рКВН+ = 5,8. При рН 5,8 в растворе в равных концентрациях находятся молекулярная и катионная формы бензимидазола, а при рН 4,8 концентрация катионной формы в 10 раз превышает концентрацию молекулярной. Для эквивалентного обмена катионов необходимо поддержание высокой концентрации катионной формы БИА и электролит должен быть слабокислым. При этом резко уменьшается скорость ЭОП, а также возрастает общая концентрация электролита, что приводит к возрастанию тока в капилляре. На практике ведущий электролит готовят на основе винной кислоты, анионы которой обладают малой подвижностью и увеличивают сопротивление электролита, а соотношение кислоты и основания подбирают до достижения необходимого компромисса между временем анализа и величиной тока.
При электрофорезе катионы регистрируются согласно их электрофоретической подвижности. Первыми мигрируют NH4+ и K+, их подвижности одинаковы и обычно они выходят одним пиком. Для разделения NH4+ и K+ в состав ведущего электролита вводят макроцикл 18-краун-6 с гидрофильной внутренней полостью, размер которой соответствует ионному радиусу К+. В результате образуется комплекс включения «гость»–«хозяин». В основе комплексообразования лежат ион-дипольные взаимодействия катиона К+ с атомами кислорода (рис. 8). Благодаря образованию комплекса подвижность К+ снижается, а подвижность других ионов остается без изменений (рис. 9).
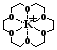
Рис. 8. Комплекс катиона калия с макроциклом 18-краун-6.
Далее с хорошим разрешением идут пики Na+, Li+, Mg2+, Sr2+, Ba2+ и Ca2+.
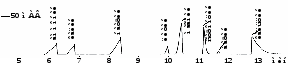
Рис. 9. Электрофореграмма модельного раствора катионов.
«Капель-105», капилляр: внутр. диаметр 75 мкм, Lэфф/Lобщ = 50/60 см.
Ведущий электролит: 10 мМ БИА, 5 мМ винная кислота, 2 мМ 18-краун-6.
Ввод пробы: гидродинамический 30 мбар*10 с. Напряжение: +13 кВ.
Температура: 20°С. Детектирование: 267 нм, косвенное.
Важно знать катионный состав дистиллированной воды, так как от этого будет зависеть возможность и достоверность определения низких концентраций анализируемых катионов (особенно NH4+).
Определение анионов (Cl , SO42, NO2, NO3, F , HPO42)
Для определения анионов в приборе «Капель» необходимо установить источник высокого напряжения отрицательной полярности. Тогда электрод на входном конце капилляра будет катодом, а электрод выходного конца анодом, и анионы будут мигрировать в сторону выходного конца, т. е. к детектору.
На рис. 10 а) показано направление движения ЭОП в противоположную от анионов сторону. Скорость движения анионов заметно превосходит скорость течения жидкости в капилляре, тем не менее, разнонаправленные потоки могут существенно увеличивать времена анализа анионов. Для использования транспортной функции ЭОП, который только переносит зоны разделенных компонентов, не принимая участия в самом процессе разделения, принято обращать направление движения ЭОП (рис. 10б), вводя в состав катионные ПАВ.
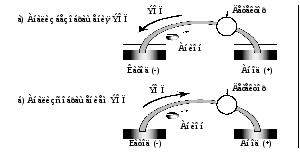
Рис. 10. Схемы анализа неорганических анионов.
Ведущий электролит для анализа анионов должен удовлетворять ряду условий.
Во-первых, он должен быть щелочным, так как большинство определяемых анионов существуют только в щелочных средах.
Во-вторых, основой электролита должен быть анион, имеющий сильную полосу поглощения в области 254 нм, так как большинство анионов не обладают собственными полосами поглощения в указанной области, и их определяют только косвенным методом.
В-третьих, ведущий электролит должен содержать добавку для обращения направления электроосмотического потока, так как в противном случае ЭОП, направленный к катоду, резко замедлит или сделает невозможной электромиграцию анионов к детектору.
В-четвертых, катионный компонент ведущего буферного раствора должен быть катионом достаточно сильного основания с малой подвижностью, чтобы обеспечить малую электропроводность раствора.
На практике рабочий буферный раствор состоит из смеси диэтаноламина (основание) и хромовой кислоты с добавкой катионного ПАВ бромида (лучше гидроксида) цетилтриметиламмония (ЦТАБ или ЦТАОН). Избыток диэтаноламина (ДЭА) создает слабо щелочную среду (рН 9), анион CrO42 обеспечивает необходимое светопоглощение, а катион ЦТА+, сорбируясь на поверхности кварцевого капилляра, перезаряжает поверхность на положительную, чем достигается изменение направления ЭОП.
Бромид цетилтриметиламмония [C16H33(CH3)3N]+Br легко растворим в воде. Как катионное ПАВ, ЦТАБ в малых концентрациях образует истинные растворы, а при более высоких коллоидные. Частицы коллоидного раствора – мицеллы – представляют собой сферические образования, состоящие из 60–100 катионов, обращенных азотным концом наружу, которые несут соответствующий положительный заряд, нейтрализуемый эквивалентным количеством анионов. ККМ для ЦТАБ равна 7 ммоль/л.
Порядок миграции анионов: Cl , NO2, SO42, NO3, F , HPO42. Все пики разрешаются полностью. Выход пика гидрокарбоната, который всегда присутствует в буферном растворе и в растворе пробы, может служить сигналом для окончания анализа.
На электрофореграммах стандартных растворов и растворов проб часто наблюдаются отрицательные пики. Их появление связано с тем, что в растворах проб (стандартов) отсутствуют анионы, которые находятся в растворе ведущего электролита. Первый такой пик может наблюдаться между пиками Cl и NO2 и связан с присутствием в составе ведущего электролита анионов Br (для ЦТАБ в качестве модификатора ЭОП, рис. 11а). Величина этого «бромидного провала» пропорциональна общей концентрации анионов в пробе, и затрудняет автоматическую разметку пика нитрита (исправляется вручную).
Второй отрицательный пик часто наблюдается после пика HPO42. Его появление объясняется тем, что при хранении буферные растворы поглощают все большие и большие количества СО2. В ряде случаев концентрация НСО3 в пробе может оказаться меньше, чем в ведущем электролите, и тогда на электрофореграмме на месте пика НСО3 появляется отрицательный пик. В особых случаях он может быть настолько большим, что будет мешать автоматической разметке пика HPO42.
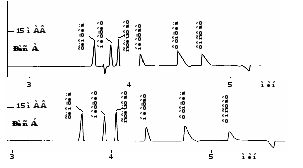
Рис. 11. Электрофоретическое разделение неорганических анионов
в присутствии (а) и в отсутствие (б) Br в составе ведущего электролита.
Это является сигналом к тому, чтобы заново приготовить свежий раствор ДЭА (быстрее всех поглощает СО2) или полностью заменить компоненты ведущего электролита на свежеприготовленные. Количественное определение НСО3 невозможно из-за его неконтролируемого содержания в используемых растворах.