хроматография-лекции. Хроматографические методы. Общая характеристика методов
 Скачать 6.82 Mb. Скачать 6.82 Mb.
|

|
Параметр h Если проанализировать влияние возможных отклонений температуры колонки и скорости потока газа-носителя от средних значений, соизмеримых по длительности с продолжительностью регистрации пика на устойчивость и линейность параметра h, то получим следующее:
Линейная связь между количеством введенной пробы и параметром h нарушается, если вводимые количества пробы существенно больше допустимых. Таким образом, при использовании в качестве параметра пика абсолютного значения h необходимо строго стабилизировать усло-вия анализа. Высота пика h является наиболее легко измеряемым параметром, и по этой причине случайные ошибки, связанные с измерениями на хроматограмме, как правило, минимальны. Параметр h рекомендуется применять в следующих случаях:
Параметр h не рекомендуется применять в случаях:
Параметр hl Влияние изменения условий процесса хроматографического разделения на параметр hl сказываются следующим образом:
туации скорости потока газа-носителя влияет на абсолютное значение величины l, поскольку эта величина зависит от скорости потока газа-носителя в момент десорбции компонента из колонки. Однако это влияние очень мало, поскольку связано с изменением l за счет изменения ширины пика у основания b. Поскольку чаще всего l >> b, этим влиянием можно пренебречь. Линейность параметра hl имеет те же ограничения, что и линейность параметра h. При использовании параметра hl, как и при любых количественных измерениях, требуется калибровка детектора по индивидуальным соединениям. Следует отметить, что по различным причинам эти калибровочные коэффициенты отличаются от коэффициентов, полученных при использовании площадей пиков. Очень важно, что калибровочные коэффициенты, определенные по hl, сохраняют свое значение в довольно широком диапазоне колебания условий анализа. Измерения, необходимые для вычисления параметра hl, легко выполнимы и хорошо воспроизводятся. Параметр hl рекомендуется использовать в следующих случаях:
Параметр hl не рекомендуется использовать в случаях:
Расчеты без применения калибровочных коэффициентов при использовании этого параметра могут давать очень большие ошибки. Использование калибровочных коэффициентов, взятых из литературы для количественных расчетов, также приводит к ошибкам. Параметр А Влияние изменения условий хроматографических разделений на параметр А (площадь пика) сводятся к следующему:
Площадь пика для детекторов, используемых в количественной хроматографии, является универсальным параметром, применимым во всех случаях, когда отсутствуют необратимые процессы в колонке и когда детектор работает в области нормальных рабочих условий. Применение потоковых детекторов при использовании параметра А предпочтительнее. Имеются сведения о количественной характеристике устойчивости различных параметров пика. Так, изменение относительной высоты пика составляет 0.30.9 % при изменении температуры на 1 К. Точность 2 % при анализе методом абсолютной калибровки по высотам пиков может быть достигнута при стабилизации температуры с точностью до 0.1 К и скорости газа-носителя до 1 мл/мин. Трудно получить результаты с относительной погрешностью менее 2.8 3.2 % при концентрации компонента в пробе 12 %, если в качестве параметра пика используется его высота. Для детектора по теплопроводности в сочетании с измерением площади пика получены следующие значения допустимых погрешностей задания параметров (при условии получения погрешности результата не более 1 %): Т а б л и ц а 12 Величины допустимых погрешностей задания параметров разделения
Эти данные получены с учетом влияния каждого фактора на конечный результат анализа. При этом вклад в погрешность результата всех шести источников частных погрешностей предполагался одинаковым. Если, например, измерять не площадь пика, а высоту пика, то допустимая погрешность изменения температуры оказывается в 2 раза меньшей. Большой вклад в погрешность вносит давление на выходе из колонки. Поскольку эта величина определяется изменением атмосферного давления, ее колебания могут быть достаточно велики (до 3 %). Между тем, в практической хроматографии это часто не учитывается. В результате возникают существенные погрешности при использовании для расчетов абсолютных значений параметров. По этой причине детектор ионизации в пламени считается более пригодным для количественных измерений, чем детектор по теплопроводности. Для получения погрешности менее 1 % при определении времени удерживания требуются не столь жесткие условия по стабильности аппаратуры: давление на входе в колонку можно поддерживать с точностью 0.4 %, температуру с точностью 0.5 К и выполнять измерения величины площади с точностью 0.5 %. 5.2 ИЗМЕРЕНИЕ КОЛИЧЕСТВЕННЫХ ПАРАМЕТРОВ ХОРОШО РАЗДЕЛЕННЫХ ПИКОВ 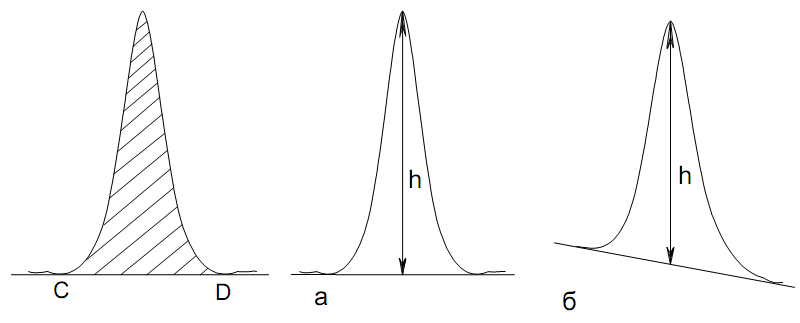 Количественный газохроматографический анализ основан на зависимости между площадью S (или высотой h) пика и количеством определяемого компонента в пробе. Под площадью пика понимается площадь, ограниченная контуром пика и его основанием — СD отрезком, соединяющем начало и конец пика (рис. 24). Площадь пика измеряется в мм2, А⋅с или В⋅с. Высота пика (h) — расстояние от вершины пика до его основания, измеренное в направлении параллельной оси сигналов детектора (рис. 25), h – измеряется в мм: а – нулевая параллельная оси времени, т.е. дрейф отсутствует (рис.25, а); б – измерение высоты при дрейфе нулевой (рис. 25, б). Площадь и высота пика могут измеряться как вручную, так и автоматически. При ручных способах измерения площади пика используют вспомогательный параметр - ширина пика (рис. 26). Ширина пика - проекция отрезка прямой, параллельной основанию пика, ограниченного точками пересечения с ветвями пика, на ось времени. Ширина пика может измеряться на разных уровнях (сечениях) высоты пика. Ширина пика измеряется в мм; сек. Наиболее просто вручную измеряются и рассчитываются параметры так называемых гауссовых пиков (рис. 27). Контур этих пиков описывается уравнением: где х, у – координаты точки контура пика; h и S – высота и площадь пика, отвечающая максимальной концентрации компонента в зоне; σ – стандартное отклонение, которое отвечает ширине пика на высоте 0,882 h. 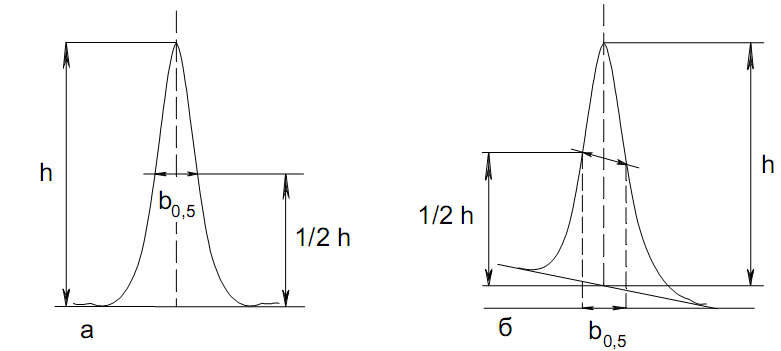 Стандартное отклонение может быть определено также из соотношений, справедливых для гауссовских пиков: 2σ = b0,607 2,36σ = b0,500 3σ = b0,324 4σ = b0,134 Расчет площади пика как площади, ограниченной гауссовой кривой (для симметричных пиков), проводят по формуле, полученной интегрированием гауссовой функции распределения ошибок:  (62) (62)где S – площадь пика; h – его высота; σ – стандартное отклонение, равное ширине пика на высоте b0,882. Точность измерения площади пика этим методом определяется точностью измерения отрезков на хроматограмме (h и b0,882), а, поскольку, высота пика обычно много больше ширины пика, то точность измерения площади пика в значительной степени определяется точностью измерения ширины пика. Поэтому при измерении ширины узких пиков (меньше 10 мм) желательно пользоваться измерительной лупой и учитывать толщину линии. 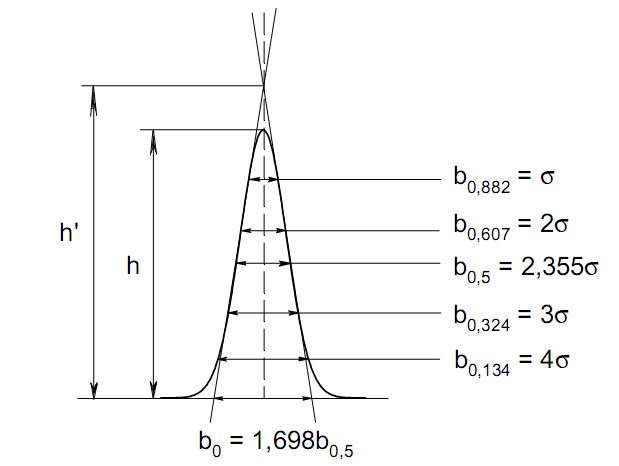 Обработка хроматограмм с асимметрическими пиками, как правило, проводится с меньшей точностью. Асимметричность пика анализируемого соединения может быть вызвана двумя причинами: 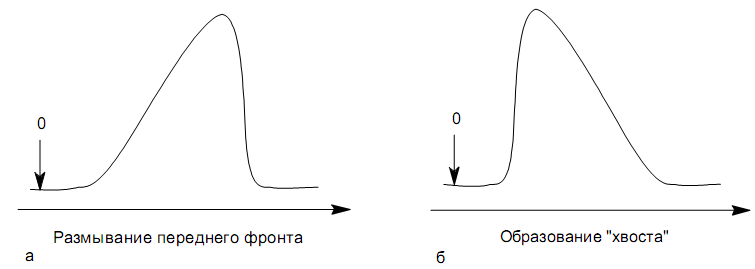 1. Перегрузкой колонки анализируемым веществом. 2. Наличием остаточной адсорбционной активности твердого носителя, используемого для приготовления сорбента. Форма пиков при этом различна. Перегрузка колонки приводит к образованию размытого фронта (рис. 28, а). Остаточная адсорбционная активность приводит к образованию хвоста (рис. 28, б). Устранение асимметричности первого рода достигается при уменьшении дозы. В то время как устранение асимметричности второго рода более трудная задача. Для численного выражения асимметричности предложено использовать коэффициент асимметричности (рис. 29): Kas=SA/SB=c/d=a/b (63) где SA и SB – площади между осевой линией пика и задним его фронтом, и передним его фронтом соответственно; а и b – расстояния между осевой линией пика и задним его фронтом и передним его фронтом на полувысоте соответственно; c и d – то же самое на 0,1 высоты пика. 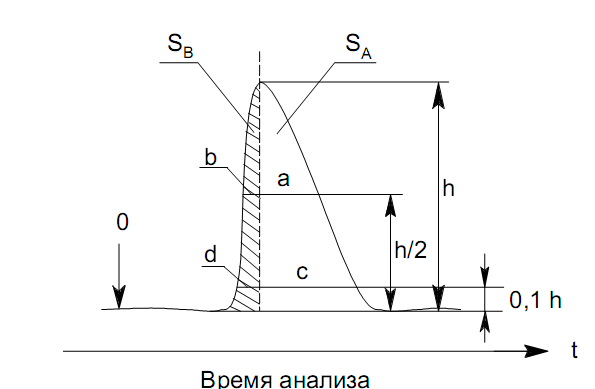 Для симметричного пика Каs = 1. В случае остаточной адсорбции Каs > 1, при «перегрузочных» пиках Каs < 1. Считается допустимым работать на колонке, имеющей Кas для всех компонентов анализируемой смеси в пределах 0,7 – 1,5. Искажение формы пика при адсорбции («хвостование») зависит и от природы анализируемых соединений. Менее всего адсорбируются насыщенные углеводороды, наиболее сильно - высокополярные соединения. Аналитическая колонка может оказаться пригодной для анализа соединений одного класса и совершенно не пригодной для соединений другого класса. Ее следует охарактеризовать и по этому признаку. 5.3.1 Методы триангуляции Пик рассматривают как треугольник и площадь его рассчитывают как площадь треугольника. Известны три метода триангуляции (triangle – треугольник). Эти методы приближенные, поскольку площадь пика аппроксимируется площадью одного из трех треугольников. а). В первом методе триангуляции за площадь пика принимается площадь описанного треугольника, образованного касательными, проведенными к сторонам пика в точках перегиба, и отрезком, отсекаемым касательными на основании пика (рис. 30, а). Площадь описанного треугольника рассчитывается обычно по формуле: S = ½ ⋅ h' ⋅ b0 (65) 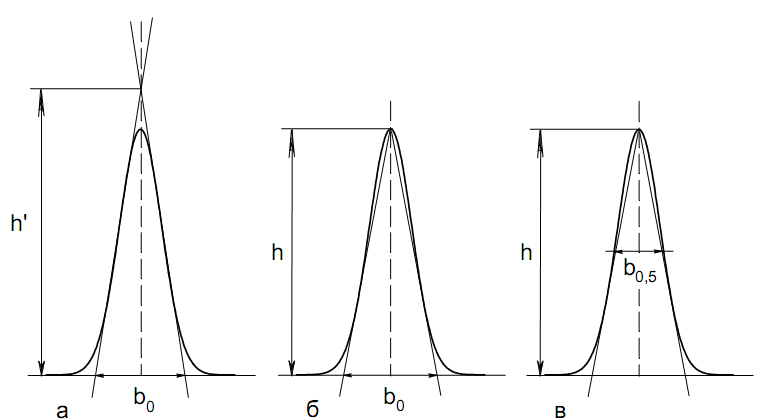 где h' – высота описанного треугольника; b0 – ширина пика на нулевой линии. Этот метод дает около 97% (96,8%) от площади гауссова пика. Недостатки этого метода состоят в необходимости дополнительных геометрических построений (проведение касательных), которые не всегда могут быть выполнены легко и с достаточной точностью. Особенно это касается узких пиков. При этом точка пересечения касательных может оказаться за пределами диаграммной ленты. б). Во втором методе триангуляции (рис. 30, б) предложено экстраполировать «прямолинейные» участки ветвей пика только вниз до пересечения с основанием и площадь рассчитывать по формуле: S = ½ ⋅ h ⋅ b0 (66) Этим методом находят около 80% (79,8%) от площади гауссового пика. Здесь частично преодолеваются недостатки первого метода. в). На практике наибольшее распространение получил третий метод триангуляции (рис. 30, в). Метод произведения высоты на ширину пика на половине его высоты. В этом случае геометрические построения упрощаются. Процесс измерения состоит из четырех операций, а именно: – проведение основания под пиком (интерполирование нулевой линии между началом и концом пика); – измерение высоты пика; – нахождение середины высоты; – измерение ширины пика на половине высоты. Площадь рассчитывается по формуле: S = h ⋅ b0,5 (67) Этим методом находят около 94% (93,9%) от площади гауссова пика. Точность измерения площади в методе «h ⋅ b» зависит от формы и абсолютных размеров пика. Форму пика принято характеризовать отношением «h / b0,5» . Ошибка минимальна при h / b0,5 равна 5−6%. С повышением абсолютных размеров пика точность измерения площади пика возрастает. Истинная же площадь гауссова пика может быть найдена по формуле: Sист = h⋅b0,368 (68) 5.3.4 Определение площадей не полностью разделенных пиков Рассмотрим случай наложения двух гауссовых пиков, когда огибающая имеет минимум (рис. 32). При наложении двух пиков происходит искажение измеряемых параметров пика (высоты и ширины). При этом степень искажения параметров зависит от полноты разделения и соотношения высот соседних пиков. Известны различные приемы расчета хроматограмм с не полностью разделенными пиками. Метод Бартлета и Смита (корректировки высоты) Площадь неразделенных пиков рассчитывается по обычной формуле для гауссовых пиков, например по формуле (73). Однако в формулу подставляют значения истинных параметров пиков, а не измеренных линейкой на хроматограмме. Корректировка высоты пиков, учитывающая их взаимное влияние. 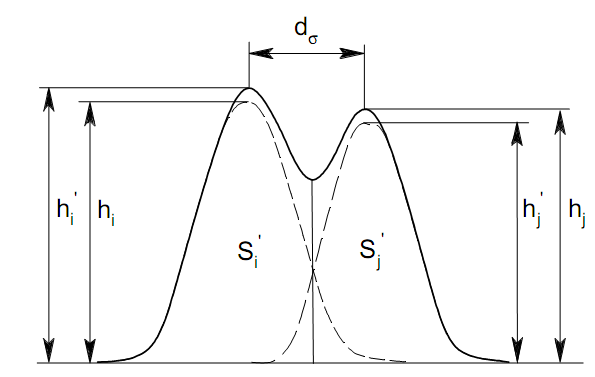 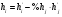 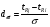 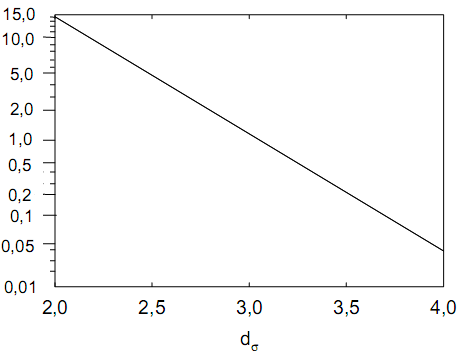 Метод опускания перпендикуляра При взаимном наложении двух пиков площадь под огибающей кривой равна сумме площадей под индивидуальными пиками (рис. 26). В этом методе границей двух не полностью разделенных пиков является перпендикуляр, опущенный из минимума огибающей кривой на основание пиков. За площадь первого пика принимается площадь, лежащая слева от перпендикуляра, а за площадь второго пика принимается площадь, лежащая справа. Этот метод приближенный, поскольку перпендикуляр является истинной границей только в случае неразделенных пиков одинаковой высоты и ширины. Для того чтобы исключить систематические ошибки, измеренные площади умножают на значения поправочных коэффициентов. В теории хроматографического анализа следует сделать два допущения: Во-первых, предполагается идентичность состава пробы, введенной в хроматограф, и смеси анализируемых веществ. Это можно выразить равенством массовых долей определяемого компонента. 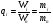 (78) (78)где Wi и mi – масса определяемого компонента в отобранной пробе анализируемых веществ и дозируемой пробе соответственно; Wn и mn – навеска анализируемой пробы и хроматографирумой пробы, соответственно. Во-вторых, предполагается линейная зависимость между параметрами пика и содержанием компонента в пробе: mi = kai ⋅ Si , (79) где kai – коэффициент пропорциональности (абсолютный калибровочный коэффициент); Si – площадь пика. В линейном динамическом диапазоне детектора ki величина постоянная и от величины пробы не зависит. Объединяя предыдущие два уравнения, получаем: 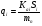 (80) (80)Итак, для определения состава анализируемой смеси необходимо учитывать различную чувствительность детектора к разным веществам, измерить количественный параметр пика и учесть массу пробы. Различные методы расчета состава смесей по хроматограммам, как будет видно из дальнейшего изложения, отличаются способом учета величины пробы. 20 40 0 5 10 15 10 30 50 % вес S, мм2 0 Известны четыре основных метода расчета состава смеси по хроматограммам: метод абсолютной калибровки, метод внутренней нормализации, метод внутреннего стандарта и метод стандартной добавки. Метод абсолютной калибровки основан на том, что для, количественного определения содержания компонентов в анализируемой пробе, необходимо для каждого определяемого в пробе индивидуального вещества построить калибровочную кривую зависимости площади или высоты хроматографического пика от содержания этого вещества в анализируемой пробе. Необходимость такой трудоемкой работы обусловлена тем, что чувствительность детектора хроматографа к различным веществам даже близкой химической структуры, как правило, неодинакова. Построение калибровочных кривых производят по результатам хроматографирования в одинаковых условиях известных количеств определяемого вещества. Зависимость площади Si или высоты хроматографического пика от содержания qi вещества в анализируемой пробе при достаточно строгом воспроизведении условий хроматографирования является линейной и угловой коэффициент (тангенс угла наклона) калибровочной прямой i-го компонента называется калибровочным коэффициентом Ki, г/см2: 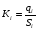 (1.59) (1.59)Процентное содержание i-го компонента Xi в пробе составит 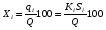 (1.60) (1.60)где Q масса анализируемой пробы, г. Метод абсолютной калибровки достаточно прост, но точность его в значительной мере зависит от постоянства режима и тщательности приготовления и анализа эталонов или их смесей. Недостаток метода состоит в том, что приходится строить калибровочные графики для каждого индивидуального вещества, на что тратится много времени и расходуются эталонные вещества. Поэтому его применяют, главным образом, при определении одного или нескольких компонентов смеси, например, микропримесей. Метод внутреннего стандарта (метод метки) основан на сравнении высот или площадей пиков известного вещества-метки и определяемых компонентов. В качестве внутреннего стандарта (метки) стараются подобрать такое вещество, которое бы не реагировало с компонентами смеси, не очень сильно сорбировалось и появлялось на хроматограмме отдельно от других компонентов. Кроме того, его не должно быть в составе исследуемой смеси. К анализируемой пробе добавляют точно известное количество вещества-метки. Количество метки подбирают такое, чтобы площадь его пика была соизмерима с площадью пиков компонентов смеси, подлежащей количественному определению. Для калибровки следует провести хроматографический анализ ряда смесей вещества-метки с каждым из отдельных определяемых компонентов при различных соотношениях обоих веществ в смеси. По оси абсцисс откладывают значения отношения массы метки qm к массе анализируемого вещества qi (qm /qi) в искусственных смесях, а по оси ординат соответствующее им соотношение площадей пиков Sm/Si. В узкой области соотношений qm/qi график прямолинеен. Угловой коэффициент такого графика Кi представляет собой калибровочную константу для данного определяемого компонента. Он вычисляется из уравнения 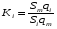 . (1.61) . (1.61)Отсюда 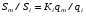 . .Калибровочная константа практически не зависит от количества добавляемой в колонку искусственной смеси и мало изменяется при изменении скорости потока подвижной фазы и температуры колонки. Относительное содержание вещества-метки Imв смеси рассчитывается по следующей формуле: 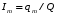 . (1.62) . (1.62)Относительное содержание определяемого компонента Ii 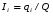 , (1.63) , (1.63)где 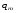 масса вещества-метки в пробе; масса вещества-метки в пробе; 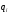 масса определяемого вещества в пробе; Q весовое количество пробы смеси, введенное в колонку. масса определяемого вещества в пробе; Q весовое количество пробы смеси, введенное в колонку.Разделив Im на Ii получаем 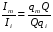 ,. (1.64) ,. (1.64)откуда 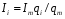 . .Процентное содержание определяемого вещества вычисляется из уравнения 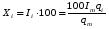 , (1.65) , (1.65)а так как 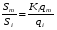 , то , то 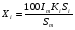 . (1.66) . (1.66) Таким образом, чтобы провести анализ и количественный расчет методом метки, нужно: 1) составить ряд искусственных смесей определяемого вещества с веществом-меткой, снять ряд хроматограмм при режиме анализа смеси и измерить соотношение площадей соответствующих пиков; на основании полученных данных построить графики в координатах 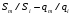 ; рассчитать калибровочную константу Ki; ; рассчитать калибровочную константу Ki; 2) определить приблизительное весовое количество анализируемой смеси, которое нужно вводить в колонку, чтобы получить достаточно хорошо выраженный пик определяемого компонента; подобрать такое количество вещества-метки, при котором на хроматограмме появился бы пик метки, соизмеримый с пиком определяемого компонента; приготовить стандартную смесь метки и компонентов; вычислить Im по следующей формуле: 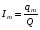 . (1.67) . (1.67)3) в хроматографическую колонку ввести пробу анализируемой смеси с известным содержанием метки и снять хроматограмму; измерить площадь пика определяемого компонента Si и пика вещества метки Sm; зная калибровочную константу Ki, рассчитать процентное содержание компонентов по формуле (1.68)Если необходимо определить несколько компонентов, то для каждого из них находят калибровочную константу описанным способом. Метод метки удобен тем, что на результаты анализа колебания параметров хроматографического опыта сказываются слабо. Но если дозировка объемов анализируемых образцов проводится достаточно точно, этот метод никаких преимуществ перед методом абсолютной калибровки не имеет. Кроме того, использование метода метки ограничивается преимущественно определением лишь небольшого числа компонентов, содержание которых в не очень сложных по составу смесях не превышает примерно 10%. |
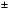 0.10 %
0.10 %