хромосомные болезни. Хромосомные болезни
 Скачать 218.64 Kb. Скачать 218.64 Kb.
|

|
Министерство образования и науки Российской Федерации (МИНОБРНАУКИ РОССИИ) Первый Московский государственный медицинский университет имени И. М. Сеченова (Первый МГМУ им. И. М. Сеченова) кафедра ботаники РЕФЕРАТ по дисциплине (по выбору) «Геном человека» на тему «Хромосомные болезни» Автор: студент группы № 2 II курса заочного отделения фармацевтического факультета ПМГМУ им. И.М. Сеченова Цвиркун Артем Николаевич Москва 2014 Содержание: Введение…………………………………………………………………………...3 Глава 1. Хромосомные болезни 1.1. Хромосомы: строение, классификация, нормальный хромосомный набор человека……………………………………………………………………………4 1.2. Виды хромосомных мутаций и причины их возникновения……………...5 1.3. Транслокации хромосом……………………………………………………..8 Глава 2. Примеры наиболее частых хромосомных патологий 2.1. Некоторые общие черты в клинике хромосомных заболеваний……..….10 2.2. Примеры наиболее частых хромосомных патологий…………………….11 2.3. Микроделеционные и дупликационные синдромы………………………14 Заключение……………………………………..………………………………..16 Выводы…………………………………………………………………………...18 Список литературы………………………………………………………………19 Введение Хромосомные болезни и синдромы достаточно хорошо изучены клинически, для многих из них идентифицированы аномалии хромосом. Однако конкретные механизмы развития заболеваний остаются предметом активного исследования, и геномный уровень анализа их патогенеза открывает новые факты. Изменение нормального числа или структуры хромосом у человека — хромосомные мутации, называемые еще хромосомными аберрациями, являются причиной развития различных заболеваний и аномалий — хромосомных болезней. Знания о хромосомных болезнях начали накапливаться с 1959 г., когда было установлено, что известная и широко распространенная болезнь Дауна, являющаяся по существу хромосомным заболеванием, возникает вследствие трисомии по хромосоме 21. С того времени и до наших дней описаны десятки новых хромосомных болезней. Хромосомные болезни классифицируются по характеру хромосомной мутации и их происхождению — наследственному или врожденному. Хромосомные мутации — это изменения числа и структуры хромосом. Изменения числа хромосом связаны с нарушением механизма клеточного деления. Они могут изменяться по числу, кратности n (гаплоидный набор Зn, 4n, 5n), это так называемые, полиплоидные геномные мутации. Глава 1. Хромосомные болезни 1.1. Хромосомы: строение, классификация, нормальный хромосомный набор человека Двухцепочечная молекула дезоксирибонуклеиновой кислоты (ДНК), компактизуясь с помощью специальных белков гистонов, сначала образует нуклеосомы, представляющие собой, как показано на рисунке 2, комплексы из 8 гистоновых молекул с накрученной на них нитью ДНК. В дальнейшем, при участии дополнительных белков нуклеосомная нить упаковывается ещё более плотно, и формирует хроматиновую нить, которая, в свою очередь, многократно складывается и образует хромосому. Наиболее хорошо хромосомы становятся видны в период деления клетки, когда хроматин максимально компактизован. Так, на стадии метафазы все хромосомы клетки могут быть четко визуализированы под микроскопом, именно это свойство активно используется в процессе проведения цитогенетического анализа – кариотипирования. 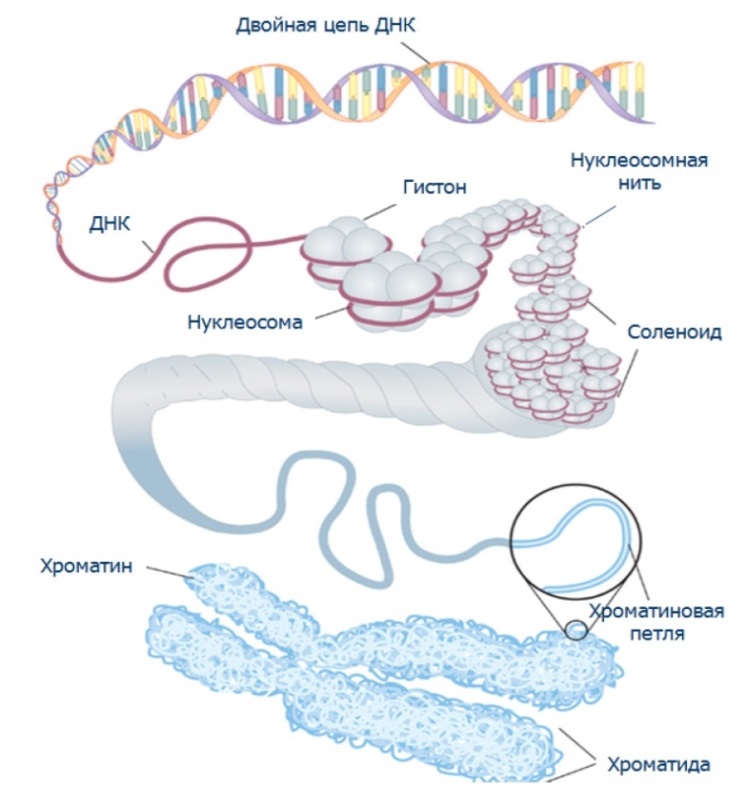 Рис.1 Организация наследственной информации, структура хромосом Хромосомы состоят из двух плеч и центромеры. Концевые участки плеч называют теломерами. Короткое плечо хромосомы принято обозначать латинской буквой p, длинное плечо - буквой q. Если по длине плечи p и q практически неотличимы, то такие хромосомы относят к группе метацентрических. В случае заметной разницы между коротким и длинным плечом – к субметацентрическим. Если p плечо практически не выражено, то – к акроцентрическим хромосомам. Хромосомный набор обозначается термином кариотип. При этом 46 хромосом представляют собой 23 пары: две первых, две вторых и т.д. Одну представительницу каждой пары мы в своё время получаем от матери, а другую от отца. Также и мы в норме передаём лишь одну представительницу каждой пары нашим будущим детям. 1.2. Виды хромосомных мутаций и причины их возникновения Рис. 2 Классификация хромосомных мутаций Хромосомные мутации могут быть подразделены на качественные и количественные. К качественным - относят делеции (потери участка хромосомы), дупликации (удвоение какого-либо локуса), инверсии (поворот участка на 180º) и транслокации (обмен локусами между разными хромосомами) (рис. 3). Обо всех этих изменениях речь пойдёт чуть позже. А пока остановимся на количественных мутациях. Как следует из их названия, они характеризуются изменением числа хромосом. Нарушение числа копий целого хромосомного набора, когда аномальный кариотип отличается изменением количества хромосом, кратным гаплоидному, называется моно- либо полиплоидией. При этом моноплоидия - это кариотип, состоящий из одного гаплоидного набора (1n). Полиплоидия - увеличение количества хромосом до 3n (триплоидия), 4n (тетраплоидия) и т.д. Подобные изменения хромосомного набора практически не встречаются среди живорождённых детей, т.к. приводят к серьёзнейшим нарушениям эмбриогенеза и чаще всего являются причиной внутриутробной гибели эмбриона на ранней стадии его развития. В свою очередь, нарушение числа хромосом, при котором мутантный организм отличается от нормального лишь какой-то частью хромосомного набора, называется анеуплоидией. Если происходит потеря одной хромосомы, то такое явление носит название моносомии. Пожалуй, в качестве одного из немногих примеров среди живорожденных детей в данном случае можно привести синдром Шерешевского-Тернера, при котором кариотип пациентки отличается отсутствием одной из Х-хромосом (45,X). В случае если в хромосомном наборе появляется дополнительная копия определённой хромосомы, такое явление носит название трисомии или полисомии. При этом трисомия – это кариотип, характеризующийся одной дополнительной, а полисомия - двумя и боле дополнительными хромосомами. Примеров хромосомных болезней с подобным изменением кариотипа гораздо больше. Это и синдром Дауна (трисомия по 21-ой паре), и синдром Патау (трисомия по 13-ой паре), и синдром Клайнфельтера (полисомия по Х-хромосоме у мальчиков), и многие другие. Такое обилие примеров полисомий и практически отсутствие моносомий среди людей говорит о том, что утрата генетического материала целой хромосомы очень серьёзно сказывается на внутриутробном развитии зародыша и чаще всего, как и при моно- и полиплоидии ведёт к его внутриутробной гибели. В то же время большая часть эмбрионов, заложенных с какой-либо трисомией, также элиминируется ещё на ранних сроках беременности. По последним данным, как показало цитогенетическое исследование эмбриональных тканей, порядка 60% всех случаев самопроизвольных прерываний беременности I триместра (неразвивающиеся беременности, самопроизвольные выкидыши) связаны с той или иной хромосомной аномалией у плода. Считается, что таким образом у человека проявляется естественный отбор. Как показывает практика, в основном дети с хромосомной патологией появляются у совершенно здоровых родителей, иными словами вовсе необязательно иметь аномальный хромосомный набор для того, чтобы родить, например, ребёнка с синдромом Дауна. Причина таких случаев кроется в возникновении хромосомной мутации de novo (т.е. новой мутации) в одной из половых клеток родителей. Следует упомянуть, что спонтанные хромосомные мутации могут происходить не только на уровне половых клеток, но и в соматических клетках. Например, такая мутация может произойти под воздействием какого-либо тератогенного фактора при митотическом делении в одной из клеток эмбриона на ранних стадиях его развития. В таком случае аномальная клетка с добавочной хромосомой даст начало клону клеток с аномальным кариотипом. В результате это приведёт к рождению ребёнка с мозаичной формой хромосомной патологии, когда часть клеток организма несёт нормальный хромосомный набор, а часть – патологический. Подобные ситуации в одной семье повторяются крайне редко, поэтому риск повторного рождения в семье ребёнка с таким же хромосомным заболеванием при мозаичной форме считается низким. 1.3. Транслокации хромосом Рис.3 Типы хромосомных перестроек и их последствия Однако бывает так, что в семье фенотипически здоровых родителей возникает закономерный риск рождения ребёнка с хромосомной патологией. И связано это, как правило, с носительством одним из супругов сбалансированной транслокации хромосом. Транслокацией называется перенос генетического материала с одной хромосомы на другую. Реципрокными транслокациями считаются транслокации, при которых разрывы возникают одновременно в двух хромосомах и последние обмениваются образовавшимися свободными сегментами. Чаще всего в такую перестройку вовлекаются длинные плечи 11 и 22 хромосом, но могут быть задействованы и другие хромосомы. При этом изменяется порядок сегментов на хромосоме, но потери генетического материала не возникает, и, соответственно, фенотипически данный вид перестроек никак себя не проявляет. Такой человек прекрасно социально адаптирован, ведёт обычный образ жизни и, как правило, ничего не подозревает о том, что он является носителем хромосомной перестройки. Однако подобное изменение хромосом может приводить к образованию несбалансированных с точки зрения своего хромосомного набора гамет, последнее ведёт к закономерному риску рождения у таких людей детей с хромосомной патологией. На рис. 3 представлен особый вид реципрокных транслокаций – робертсоновская транслокация. При данном виде транслокации две акроцентрические хромосомы теряют короткие плечи, а длинные плечи сливаются друг с другом, формируя вместо двух одну химерную хромосому. В коротких плечах акроцентрических хромосом в основном локализуются гены рРНК, которые многократно дублируются в других акроцентрических хромосомах. Поэтому потеря коротких плеч акроцентрических хромосом не сопровождается какой-либо существенной симптоматикой. В данном случае в перестройке задействованы 14-я и 21-я хромосомы, что ведёт к формированию разного типа гамет, среди которых часть несёт добавочный материал 21-ой хромосомы. При оплодотворении такой яйцеклетки сперматозоидом с нормальным хромосомным набором произойдёт закладка эмбриона с так называемым транслокационным вариантом синдрома Дауна. В случае участия в робертсоновской транслокации двух 21-х хромосом, риск рождения ребёнка с синдромом Дауна у носителя перестройки достигает 100%. Глава 2. Примеры наиболее частых хромосомных патологий 2.1. Некоторые общие черты в клинике хромосомных заболеваний Хромосомные болезни выражаются в виде синдромов с множеством аномалий в развитии человека. Каждый синдром, обусловленный определенным нарушением кариотипа пораженного лица, имеет характерные симптомы, но существуют и некоторые общие особенности, типичные для каждого хромосомного заболевания. К ним относятся: а) дисморфизм, который проявляется в виде самых разнообразных конкретных изменений, но закономерен при всех хромосомных заболеваниях; б) нарушение интеллектуального развития, которое в большинстве случаев значительно отстает; в) развитие множественных аномалий скелета и внутренних органов. Таким образом, эти симптомы, независимо от разнообразия форм и степени их проявления, являются характерными для всех хромосомных заболеваний. Указанные выше общие особенности хромосомных заболеваний в сочетании с семейным анамнезом, в котором имеются данные о спонтанных абортах, о мертворожденных, о страданиях наследственными заболеваниями других членов семьи, дают серьезные основания для того, чтобы думать об их генезе и предпринимать соответствующие исследования для выявления хромосомных заболеваний. Установление диагноза хромосомного заболевания имеет большое практическое значение. Особенно важно определить — является ли оно врожденным или наследственным. Используя возможности пренатальной диагностики, следует определить нормален ли плод или имеет отклонения в кариотипе и в зависимости от этого принять решение об абортировании беременной женщины. Это позволяет ограничить рождение дефектных детей. Такие возможности ясно показывают большое социальное и медицинское значение своевременной и точной диагностики каждого хромосомного заболевания. 2.2. Примеры наиболее частых хромосомных патологий Синдром Дауна представляет собой трисомию по 21-й паре хромосом. Кариотип при простой полной форме выглядит следующим образом: 47,ХХ,+21 (если больна девочка) и 47,ХУ,+21 (если болен мальчик). Частота 1:700-1:800 новорожденных и зависит от возраста матери. Основные признаки с. Дауна у новорождённого: (6 и более признаков встречаются у 89% детей с с. Дауна; 4 - у 100%):
Формы:
Синдром Эдвардса является вторым по частоте после синдрома Дауна и представляет собой трисомию по 18-й паре хромосом. Кариотип при простой полной форме имеет следующий вид: 47,ХХ,+18 (если больна девочка) и 47,ХУ,+18 (если болен мальчик). Частота 0,3:100 новорожденных. В целом при рассматриваемом синдроме описано более 130 различных аномалий.
Синдром Патау представляет собой трисомию по 13-й паре хромосом. Кариотип при простой полной форме выглядит следующим образом: 47,ХХ,+13 (если больна девочка) и 47,ХУ,+13 (если болен мальчик). Частота 1:5000 новорожденных. Наиболее частые признаки синдрома Патау, встречающиеся с частотой более 50%.
Синдром Клайнфельтера представляет собой полисомию по Х хромосоме у мальчиков. Кариотип при классической форме выглядит следующим образом: 47,ХXY. Однако может быть и большее количество добавочных Х хромосом: 48,XXXY; 49,XXXXY. Частота 1:500 мальчиков. До периода полового созревания мальчики развиваются почти нормально, лишь с небольшим отставанием в психическом развитии (IQ 85-90). Однако при кариотипе 48,XXXY или 49,XXXXY может быть выражена умственная отсталость (IQ 20-78).
Синдром Шерешевского-Тернера представляет собой моносомию по Х хромосоме. Кариотип при простой полной форме выглядит следующим образом: 45,Х. Частота 1:2500 девочек.
2.3. Микроделеционные и дупликационные синдромы Как было упомянуто ранее, часть хромосомных болезней связана с качественным изменением хромосом, например, с появлением делеций или дупликаций в хромосомном материале. На сегодняшний день известно большое количество разнообразных наследственных синдромов, которые связывают с данным видом мутаций. Часть из них, в силу малого размера дефекта, не заметного под микроскопом при проведении кариотипирования, относят, к так называемым, микроделеционным и микродупликационным синдромам. Из табл. 1 видно, что, как и для большинства хромосомных болезней, несмотря на разнообразие клинической симптоматики, их объединяет один общий симптом - наличие у больных умственной отсталости.
Заключение В последние десятилетия многие исследователи обращались к причинам возникновения хромосомных болезней. Не вызывало сомнений, что образование хромосомных аномалий (и хромосомных, и геномных мутаций) происходит спонтанно. Экстраполировались результаты экспериментальной генетики и предполагался индуцированный мутагенез у человека (ионизирующая радиация, химические мутагены, вирусы). Однако реально причины возникновения хромосомных и геномных мутаций в зародышевых клетках или на ранних стадиях развития зародыша до сих пор не расшифрованы. Проверялись многие гипотезы нерасхождения хромосом (сезонность, расово-этническая принадлежность, возраст матери и отца, задержанное оплодотворение, порядок рождения, семейное накопление, лекарственное лечение матерей, вредные привычки, негормональная и гормональная контрацепция, флюридины, вирусные болезни у женщин). В большинстве случаев эти гипотезы не подтвердились, но генетическая предрасположенность к болезни не исключается. Хотя в большинстве случаев нерасхождение хромосом у человека спорадическое, можно предполагать, что оно в определенной степени генетически детерминировано. Об этом свидетельствуют следующие факты:
К биологическим факторам повышения риска нерасхождения хромосом относится возраст матери, хотя механизмы этого явления неясны (рис. 3). Как видно из рис.3, риск рождения ребенка с хромосомной болезнью, обусловленной анеуплоидией, с возрастом матери постепенно повышается, но особенно резко после 35 лет. У женщин старше 45 лет каждая 5-я беременность завершается рождением ребенка с хромосомной болезнью. Наиболее четко возрастная зависимость проявляется для трисомии 21 (болезнь Дауна). Для анеуплоидий по половым хромосомам возраст родителей либо совсем не имеет значения, либо его роль очень незначительна. 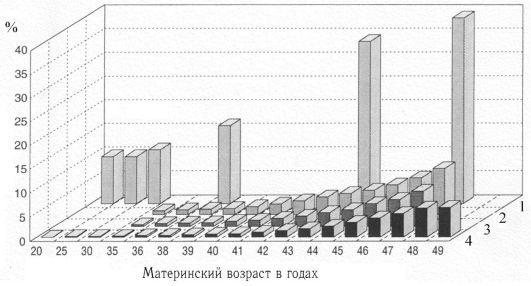 Рис. 3 Зависимость частоты хромосомных аномалий от возраста: 1 - спонтанные аборты при зарегистрированных беременностях; 2 - общая частота хромосомных аномалий во II триместре; 3 - синдром Дауна во II триместре; 4 - синдром Дауна среди живорожденных. Выводы Как было упомянуто ранее, при формировании половых клеток в норме лишь одна представительница каждой пары должна оказаться в яйцеклетке или сперматозоиде. Если по каким-либо причинам в процессе оогенеза или сперматогенеза хромосомы гомологичной пары не разошлись относительно друг друга, то образуется половая клетка, несущая дополнительную (лишнюю) хромосому. При оплодотворении такой яйцеклетки нормальным сперматозоидом будет заложен эмбрион с хромосомной аномалией. В связи с наличием хоть и небольшой, но для каждого из нас существующей, вероятности появления в половых клетках спорадических хромосомных мутаций, в настоящее время в большинстве стран мира, в том числе и в России, законодательно закреплено обязательное проведение биохимического скрининга всем беременным женщинам с целью выявления группы риска по рождению детей с хромосомной патологией. На сегодняшний день выделяют, по меньшей мере, 30 генов, ответственных за процесс расхождения гомологичных хромосом относительно друг друга. Наличие мутаций в этих генах может способствовать более частому появлению хромосомных мутаций de novo. При полной уверенности в правильности постановки диагноза хромосомной патологии по фенотипу, крайне важно провести цитогенетическое исследование. Ведь от формы кариотипа пациента будут напрямую зависеть рекомендации семье, в которой он родился. Так, если определена транслокационная форма хромосомной патологии, золотым стандартом является проведение кариотипирования родителей больного ребёнка. В этом случае велика вероятность, что один из них окажется носителем сбалансированной перестройки хромосом. При подтверждении данного факта при каждой последующей беременности семье рекомендуется проведение пренатальной диагностики. Список литературы
| ||||||||||||||||||||||||