Курсовая работа. Инулин из листьев матьимачехи. Экстракция, методы количественного и качественного анализа. Стандартизация сырья
 Скачать 296.36 Kb. Скачать 296.36 Kb.
|

|
Глава 3. Экстракция, методы количественного и качественного анализа полисахаридов мать-и-мачехи Несмотря на указание о содержании инулина в листьях мать-и-мачехи, в проанализированной научной литературе мы не обнаружили публикаций о его выделении из данного вида сырья. Вероятно, это связано с тем, что Tussilago farfara не рассматривают как промышленный источник для получения инулина. Присутствует лишь одна работа (Оленников Д.Н. с соавт.), указывающая на выделение фруктанов из листьев мать-и-мачехи [11]. Определение присутствия этого класса полисахаридов определяли следующим образом: фруктаны – положительная резорциновая реакция, выделение фракции нейтральных полисахаридов на ДЭАЭ-целлюлозе (СО32--форма), определение моносахаридного состава, 13С-ЯМР [11, 19]. Фруктаны выделяли по следующей методике. Сырье, очищенное от липофильных и фенольных компонентов несколькими растворителями, экстрагировали 80% этиловым спиртом (фракция СУ-свободных углеводов), затем водой (фракция водорастворимых полисахаридов - ВРПС). Содержание фруктозы во фракциях определяли модифицированным резорциновым методом. Ионообменная хроматография на ДЭАЭ-целлюлозе и гель-хроматография на Molselect G-25 и Sephacryl S-300 HR были использованы для выделения домининирующего полимера в составе полисахаридной фракции. Определение моносахаридного состава проводили после гидролиза полисахаридной фракции с ТФУ (в течение 2 ч при нагревании 120°С) методом ВЭЖХ [19]. Cпектрофотометрическая методика определения содержания полифруктанов сырье, использующая модифицированный резорциновый метод определения кетоз, представлена ниже [13]. В основе методики – проба Селиванова – реакция обнаружения фруктозы (и других кетоз), заключающаяся во взаимодействии фруктозы с резорцином в присутствии концентрированной соляной кислоты. Появляется вишнево-красное окрашивание. Альдозы в этих условиях взаимодействуют медленнее и дают бледно-розовую окраску или вообще не взаимодействуют.  Оленников Д.Н. с соавт. разработали методику количественного определения фруктозы в растительном сырье и в выделяемых из него углеводных комплексах при исследовании этой колориметрической реакции, подобрав оптимальные условия её проведения, используя тиомочевину для стабилизации окрашенного комплекса фруктозы с резорцином [12]. Около 1 г (точная навеска) измельченного сырья помещают в колбу со шлифом вместимостью 250 мл, приливают 100 мл 95% спирта этилового, присоединяют обратный холодильник и нагревают на кипящей водяной бане в течение 60 мин. После охлаждения извлечение фильтруют. Экстракцию сырья повторяют в тех же условиях еще 2 раза со 100 и 50 мл экстрагента. К обработанному сырью приливают 100 мл воды очищенной и нагревают на кипящей водяной бане в течение 60 мин. Извлечение фильтруют в мерную колбу вместимостью 250 мл. Экстракцию сырья повторяют в тех же условиях еще 2 раза со 100 и 50 мл воды очищенной. Объем объединенного фильтрата доводят водой очищенной до метки (раствор А) [13]. 1 мл раствора А переносят в пробирку вместимостью 25 мл, приливают 1 мл 0,01% раствора тиомочевины, 1 мл 1% раствора резорцина, 8 мл 95% спирта этилового, 9 мл кислоты хлористоводородной концентрированной и нагревают на кипящей водяной бане в течение 8 мин. После охлаждения реакционную смесь переносят в мерную колбу вместимостью 100 мл и доводят объем раствора до метки водой очищенной (раствор Б). Оптическую плотность раствора Б определяют при длине волны 480 нм. Для приготовления раствора сравнения 1 мл раствора А переносят в пробирку вместимостью 25 мл, приливают 1 мл 0,01% раствора тиомочевины, 9 мл 95% спирта этилового, 9 мл кислоты хлористоводородной кон центрированной и нагревают на кипящей водяной бане в течение 8 мин. После охлаждения реакционную смесь переносят в мерную колбу вместимостью 100 мл и доводят объем раствора до метки водой очищенной. Суммарное содержание полифруктанов (Х) в пересчете на фруктозу в процентах и абсолютно сухое сырье вычисляют по формуле [13]: 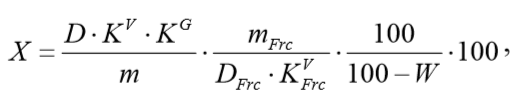 где D – оптическая плотность исследуемого раствора; DFrc – оптическая плотность раствора стандартного образца фруктозы; m – масса сырья, г; mFrc – масса стандартного образца фруктозы, г; KV – коэффициент разбавления исследуемого раствора (25000); KVFrc – коэффициент разбавления раствора стандартного образца фруктозы (10000); KG – коэффициент гидролиза (0,91); W – потеря в массе при высушивании сырья, %. Оленников Д.Н. с соавт. разработал методику количественного определения группового состава углеводного комплекса растительных объектов (свободные углеводы, водорастворимые полисахариды, пектиновые вещества и гемицеллюлозы) из одной навески, основанную на объединении известной схемы разделения углеводов по Бейли и модифицированного спектрофотометрического метода Дрейвуда [14]. Определение свободных углеводов (СУ). При выборе оптимального экстрагента для СУ необходимо было соблюдение следующих условий: растворитель должен был извлекать наибольшее количество СУ при минимальном содержании ВРПС; он должен быть доступным и нетоксичным, что имеет значение при проведении массового анализа. Около 2,0 г (точная навеска) измельченного сырья помещают в колбу со шлифом, приливают 70 мл 80%-ного спирта этилового, закрывают обратным холодильником и нагревают на кипящей водяной бане в течение 1 ч. После охлаждения извлечение фильтруют в мерную колбу вместимостью 250 мл. Экстракцию повторяют в тех же условиях еще два раза. Объем объединенного фильтрата доводят 80%-ным спиртом этиловым до метки (раствор А1). 0,5 мл раствора А1 переносят в центрифужную пробирку, приливают 0,5 мл 10%-ного раствора свинца ацетата, перемешивают и нагревают на кипящей водяной бане в течение 10 мин. После охлаждения к содержимому пробирки приливают 0,5 мл 10%-ного раствора натрия сульфата и через 20 мин центрифугируют в течение 10 мин со скоростью вращения 3000 об/мин. Надосадочную жидкость переносят в другую пробирку, приливают 4 мл 0,2%-ного раствора антрона в кислоте серной концентрированной, нагревают на кипящей водяной бане в течение 15 мин. Содержимое пробирки после охлаждения переносят в мерную колбу вместимостью 25 мл 95%-ным спиртом этиловым и доводят до метки тем же растворителем (раствор Б1). Антроновый метод основан на расщеплении сложных углеводов до моносахаридов в сильнокислой среде с последующей их дегидратацией и образованием оксиметилфурфурола, образующего при реакции с антроном комплексное соединение синевато-зеленого цвета. Интенсивность образовавшейся окраски пропорциональна содержанию сахаров в реакционной среде [17]. Определение водорастворимых полисахаридов (ВРПС). Представители этого класса углеводов вместе с ПВ относятся к так называемым биодоступным полисахаридам, так как именно они играют наиболее важную роль при развитии растения, а также отвечают за биологическую активность экстракционных препаратов из лекарственного сырья. После удаления СУ из сырья водная экстракция позволяет практически нацело извлечь этот класс углеводов. Выход ВРПС при двукратной экстракции составляет 94–97%. Остаток сырья после спиртовой экстракции извлекают водой очищенной (2×100 мл, 100 °С, 1 ч). Извлечение фильтруют в мерную колбу вместимостью 200 мл и доводят до метки тем же экстрагентом (раствор А2). 2 мл раствора А2 переносят в центрифужную пробирку, приливают 8 мл 95%-ного спирта этилового, перемешивают и нагревают на кипящей водяной бане в течение 10 мин. После охлаждения содержимое пробирки центрифугируют в течение 10 мин со скоростью вращения 3000 об/мин. Надосадочную жидкость сливают, а осадок продувают в пробирке горячим воздухом до удаления следов этанола. К осадку приливают 4 мл 0,2%-ного раствора антрона в кислоте серной концентрированной, нагревают на кипящей водяной бане в течение 10 мин. Содержимое пробирки после охлаждения переносят в мерную колбу вместимостью 25 мл 95%-ным спиртом этиловым и доводят до метки тем же растворителем (раствор Б2). Определение пектиновых веществ (ПВ). Основными типами экстрагентов для ПВ являются разбавленные растворы кислот и кислых солей, а также некоторые комплексные и хелатные соединения. Использование для этих целей растворов неорганических кислот (HCl, H2SO4, H3PO4) не позволяет добиться полного извлечения. Следует отметить, что также возрастает риск деструкции ПВ в условиях кипящей водяной бани; уменьшение температуры экстракции снижает степень разрушения ПВ с одновременным понижением выхода данной группы полисахаридов. Смесь оксалат аммония – щавелевая кислота нацело экстрагирует ПВ в щадящих условиях даже при температуре кипения (степень разрушения ПВ в течение 1,5 ч не превышает 2%). Остаток сырья после водной экстракции обрабатывают смесью 0,5%-ных растворов кислоты щавелевой и аммония оксалата (1 : 1; 3×80 мл, 100 °С, 1,5 ч) и далее по схеме для ВРПС (растворы А3 и Б3). Определение гемицеллюлоз (ГЦ). Гемицеллюлозы как группа щелочерастворимых полисахаридов хорошо извлекаются растворами неорганических оснований и основных солей. В качестве экстрагента выбран 5% раствор КОН, так как экстрагирующая сила NaOH несколько ниже. Повышение концентрации щелочи не дает особых преимуществ для извлечения ГЦ, повышение температуры снижает выход, вероятно по причине деструкции. Полнота экстракции в условиях постоянного перемешивания достигается после 4 ч; увеличение времени контакта фаз не приводит к заметному возрастанию выхода ГЦ. Остаток сырья после экстракции ПВ обрабатывают 5%-ным раствором калия гидроксида (3×80 мл, 18–21 °С, 4 ч) и далее по схеме для ВРПС (растворы А4 и Б4) – суммарное содержание ГЦ. ГЦ группы А определяют по следующей схеме: 1 мл раствора А4 переносят в центрифужную пробирку, приливают 2 мл 5%-ной кислоты серной, перемешивают и нагревают на кипящей водяной бане в течение 5 мин и далее по общей схеме (Б5). ГЦБ находят по разности значений общего содержания и ГЦА. Оптическую плотность растворов Б измеряют на спектрофотометре при длинах волн 430 (Б1, Б3–Б5) и 424 (Б2) нм в кювете с толщиной слоя 10 мм. В качестве раствора сравнения используют 4 мл 0,2%-ного раствора антрона в кислоте серной концентрированной, выдержанные в тех же условиях, что и опытная смесь. Содержание групп углеводов (Х, %) в пересчете на доминирующий моносахарид и абсолютно сухое сырье рассчитывают по формуле [14]:  где D – оптическая плотность исследуемого раствора; kV – коэффициент разбавления (12500 – СУ, 2500 – ВРПС, 5000 – ПВ, ГЦ); 0,91 – коэффициент гидролиза; E – коэффициент пересчета на моносахарид (Ara – 67, Frc – 423, Gal – 224, GalUA – 214, Glu – 358, Xyl - 455); m – масса сырья, г; W – потеря в массе при высушивании сырья, %. Разработанные подходы были применены для анализа группового состава углеводов Tussilago farfara (табл.2) [14]. Таблица 2. Групповой состав углеводов листьев мать-и-мачехи (n 11; P 0,95; tP,f 2,23)
Следует отметить, что при использовании данной методики при анализе объектов, содержащих гомополимеры типа инулина или крахмала, по показателю «водорастворимые полисахариды» наблюдается получение заниженных результатов, что объясняется частичной растворимостью этого типа полисахаридов в спирте этиловом высоких концентраций и недостаточной его преципитационной способностью. Этот факт был обнаружен еще в 30–50 гг. XX в. и описан в работах. При исключении этапа осаждения этанолом получаются более правильные результаты [14]. Детальное исследование полисахаридного комплекса (ПСК) листьев мать-и-мачехи проведено Корж А.П. [5]. ПСК выделяли из растительного сырья по разработанной методике, позволяющей получить максимальный набор полисахаридов в одном образце с минимальным количеством сопутствующих примесей растительного происхождения (рис. 4) [5].  Рис. 4. Схема выделения полисахаридных комплексов из растительного сырья Рис. 4. Схема выделения полисахаридных комплексов из растительного сырьяПолисахаридный комплекс (ПСК) выделяли из растительного сырья по следующей методике: 20 г листьев мать-и-мачехи экстрагировали раствором 2 мл концентрированной кислоты хлористоводородной в 400 мл воды очищенной (РН 4,0) при соотношении сырья и экстрагента, равном 1: 20 при нагревании на кипящей водяной бане и периодическом перемешивании в течение 3—4 ч. После отделения частиц сырья путем фильтрования через многослойный тканевый фильтр фильтрат упаривали на роторном испарителе при температуре не более 50 °С до 1/5 от исходного объема. К полученному раствору добавляли трехкратный объем 96%-го спирта этилового и отстаивали 24 ч при температуре 2—4 °С, затем осадок отфильтровывали через бумажный фильтр и растворяли в 100 мл воды очищенной при перемешивании на магнитной мешалке в течение 3 ч при комнатной температуре. Нерастворившийся остаток, представляющий собой мельчайшие частицы сырья и денатурированный белок, отделяли центрифугированием (4 000 об/мин в течение 30 мин). Центрифугат диализировали через полупроницаемую мембрану с диаметром пор 15 кДа (Orange Scientific, Бельгия) в течение 48 ч в 5 л воды очищенной (для удаления инулина (молекулярная масса от 7 до 10 к Да) и других низкомолекулярных веществ) с добавлением 0,01%-го раствора натрия азида в качестве консерванта при комнатной температуре и перемешивании на магнитной мешалке, меняя воду через 24 ч. После диализа раствор замораживали и лиофильно высушивали [7, 8]. Содержание уроновых кислот (УК) в ПСК определяли спектрофотометрическим методом по реакции с карбазолом (модификация с добавлением кислоты сульфаминовой) с применением градуировочного графика, построенного для кислоты галактуроновой [6]. Карбазол-серный метод определения галактуроновой кислоты основанн на цветной реакции карбазола с продуктами окисления моносахаров, образующихся после разрушения полимерных молекул полисахаридов концентрированной серной кислотой. Преимуществом метода является возможность избирательного определения кислых и нейтральных сахаров. Эта особенность основана на различиях в УФ-спектрах продуктов деградации нейтральных сахаров и уроновых кислот [3]. Точную навеску 0,025 г ПСК помещают в мерную колбу на 25 мл, добавляют 10 мл воды очищенной и перемешивают до растворения ПСК, далее доводят до метки водой очищенной (раствор А). В две пробирки помещают по 0,01 мл 4 М сульфаминовой кислоты. В первую пробирку прибавляют 0,25 мл раствора А (анализируемый раствор), во вторую 0,25 мл воды очищенной (раствор сравнения). Пробирки с содержимым тщательно перемешивают, затем при 0—2 °С по стенке пробирки медленно добавляют 1,5 мл раствора натрия тетрабората в серной кислоте, охлажденного до 40 °С, перемешивают, нагревают на кипящей водяной бане 6 мин, затем пробирки остужают до комнатной температуры, добавляют по 0,05 мл 0,2% раствора карбазола, тщательно перемешивают и нагревают на кипящей водяной бане 10 мин. Далее пробирки с содержимым охлаждают до комнатной температуры. Оптическую плотность анализируемого раствора измеряют относительно раствора сравнения при длине волны 535±2 нм в кювете с толщиной слоя 10 мм. Закон Бугера-Ламберта-Бера соблюдается в интервале от 0,001 до 0,05%. Определяемый минимум 0,01 мг/мл в пересчете на галактуроновую кислоту. Содержание уроновых кислот в ПСК и во фракциях (Х) вычисляют по формуле [6]:  где Сх — количество УК, найденное по градуировочному графику, мг/мл; 25, 1,81 - разведения; 0,25 — объем анализируемого раствора, взятый для анализа, мл; 0,025 — точная масса ПСК (полисахаридной фракции), г; 1000 — перевод гр. в мг. Содержание белка определяли по методу Лоури с применением градуировочного графика для бычьего сывороточного альбумина. Содержание нуклеиновых кислот устанавливали методом Спирина. Оптическую плотность растворов измеряли на спектрофотометре Unico 2800 (США) [7]. Полученный из листьев мать-и-мачехи ПСК представляет собой белую пористую массу, медленно растворимую в воде с образованием вязкого раствора. Выход ПСК составляет (3,25 + 0,05)% в пересчете на абсолютно-сухое сырье. Содержание УК в ПСК — (26,35 + 0,19), белка — (0,16 + 0,01)%, нуклеиновых кислот — (0,029 + 0,001)% [7]. Разделение ПСК на фракции проводили на колонке с DЕАЕ-целлюлозой (Sегvасеllе DЕАЕ-52, США). Навеску ПСК (0,3 г) растворяли в воде очищенной (25 мл) при перемешивании на магнитной мешалке в течение 3 ч, далее центрифугировали при 5 000 об/мин в течение 30 мин. Полученный раствор наносили на колонку (24 х 2,5 см) с DЕАЕ-целлюлозой. Колонку последовательно промывали растворами натрия хлорида с возрастающей концентрацией — 0,01; 0,1; 0,2; 0,3; 0,4; 0,5; 1,0 моль. Скорость элюента 46 мл/ч, отбирали фракции по 20 мл. Наличие полисахаридов во фракциях детектировали по положительной реакции с фенолом в присутствии концентрированной серной кислоты. Фракции концентрировали на концентраторах Vivaspin 20 (Sartorius, Германия) с диаметром пор 5 кДа до 0,5—1,0 мл при 5000 об/мин, промывая 2—3 раза водой очищенной, после чего лиофилизировали и взвешивали [7]. В результате ступенчатого элюирования растворами натрия хлорида получено 32 фракции, из которых выбирали шесть основных фракций с наибольшим выходом: РSТ-1, РSТ-2, РSТ-3, РSТ-4, РSТ-5, РSТ-6. Полученные полисахаридные фракции были охарактеризованы по содержанию белка и уроновых кислот, которое устанавливали спектрофотометрическим методом [7]. Характеристика полученных фракций представлена в табл. 3. Таблица 3. Характеристика фракций ПСК листьев мать-и-мачехи по результатам спектрофотометрического анализа
Установлено, что все фракции содержат незначительные количества белка (0,47—1,21%) (табл. 3). Минорные количества УК (от 1,13 до 4,40%), установленные во фракциях РSТ-1, РSТ-2, РSТ-3, РSТ-5, РSТ-6 (табл. 3), указывают на то, что преобладающими компонентами ПСК листьев мать-и-мачехи являются нейтральные полисахариды. Суммарный выход этих фракций от массы ПСК, нанесенного на колонку с DЕАЕ-целлюлозой, составляет 67%. Фракция РSТ-4 характеризуется высоким содержанием УК (около 58%). Выход этого полисахарида от массы ПСК, нанесенного на колонку с DЕАЕ-целлюлозой, составляет 33% [7]. Кислотный гидролиз: 3—5 мг полисахаридной фракции нагревали в запаянной ампуле 5 ч при температуре 100 °С с 1 мл кислоты трифторуксусной (ТФУ) концентрацией 2 моль, содержащей миоинозит (0,3 мг) в качестве внутреннего стандарта. Трифторуксусную кислоту удаляли многократным упариванием досуха с добавлением метанола. Получение триметилсилильных (ТМС) производных моносахаридов: к полученной в результате кислотного гидролиза смеси моносахаридов добавляли 80 мкл безводного пиридина, смесь выдерживали в сушильном шкафу при температуре 50 °С в течение 20 мин. Затем добавляли 30 мкл N-триметилсилилимидазола (Sigma-Aldrich Chemie GmbH, Германия), смесь выдерживали в сушильном шкафу при температуре 70 °С в течение 40 мин. К полученной смеси ТМС-производных добавляли 1 мл гексана, интенсивно взбалтывали. После расслоения смеси верхний слой (сумма ТМС-производных) отбирали. Полученные ТМС-производные моносахаридов анализировали на хроматографе Agilent 7890A с масс-селективным детектором Agilent 5975С, капиллярной колонкой НР5МS (30 м х 0,25 мм), газ-носитель — гелий (скорость потока 1 мл/мин). Температурный режим: 80 °С (1 мин) —> 220 °С (2 мин) —> 310 °С. Температура испарителя — 280 °С. Количество вводимой пробы — 1 мкл [7]. Изучение мономерного состава фракций методом хромато-масс-спектрометрии показало, что фракции РSТ-1, РSТ-2, РSТ-3, РSТ-4, РSТ-5 и РSТ-6 отличаются друг от друга как по природе мономерных звеньев, так и по их соотношению (табл. 4). Гидролизат полисахарида РSТ-1 состоит преимущественно из галактозы, арабинозы и рамнозы в соотношении 3,9 : 3,2 : 1, а манноза и ксилоза содержатся в минорном количестве. Гидролизат полисахарида РSТ-2 состоит из галактозы, арабинозы, рамнозы, маннозы и глюкозы в соотношении 3,6: 3,1 :2,5: 2,2: 1. Гидролизат РSТ-3 состоит преимущественно из галактозы, глюкозы и арабинозы в соотношении 3,7 : 1,5: 1,0. Таким образом, в основе структуры фракций РSТ-1, РSТ-2, РSТ-3, вероятно, лежит арабиногалактан. Указанные фракции отличаются друг от друга содержанием рамнозы, маннозы и глюкозы. Гидролизат фракции РSТ-4 состоит из рамнозы, галактозы и D-галактуроновой кислоты в соотношении 1,75 : 1,2: 1,0. Учитывая результаты анализа и опираясь на теорию о строении пектиновых веществ, можно предположить, что по химической структуре фракция РSТ-4 представляет собой рамногалактуронан I. Гидролизат РSТ-5 состоит только из рамнозы и, вероятно, представляет собой рамнан. Полисахарид РSТ-6 состоит преимущественно из галактозы и рамнозы в соотношении 2:1 и по химической структуре, вероятно, является галакторамнаном [7]. Таблица 4. Мономерный состав полисахаридных фракций листьев мать-и-мачехи по результатам хромато-масс-спектрометрии
Примечание. Gal А — кислота галактуроновая, Rha — рамноза, Ara — арабиноза, Хуl — ксилоза, Gal — галактоза, Маn — манноза, Сlu — глюкоза. Таким образом, установлено, что полисахаридный комплекс листьев мать-и-мачехи состоит из рамногалактуронана I (33%) и нейтральных полисахаридов (67%), которые представлены суммой арабиногалактана, рамнана и галакторамнана [7]. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||