биология ярыгин. Книга 1 Издание пятое, исправленное и дополненное
 Скачать 7.35 Mb. Скачать 7.35 Mb.
|

ГЛАВА 4КЛЕТОЧНЫЕИ МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ МЕХАНИЗМЫ ОБЕСПЕЧЕНИЯ СВОЙСТВ НАСЛЕДСТВЕННОСТИИ ИЗМЕНЧИВОСТИ У ЧЕЛОВЕКАЧеловек как вид является продуктом биологической эволюции. Процесс возникновения вида Homo sapiens и длительное существование его на Земле, так же как и других видов, обусловлены реализацией таких свойств живого, как наследственность и изменчивость. Важнейшим доказательством родства Человека с другими живыми организмами, населяющими Землю, является в первую очередь сходство химической и морфологической организации их наследственного материала, способа записи наследственной информации и ее использования. Большое сходство наблюдается также в спектре белков, образуемых в организме человека и высших животных, например человекообразных обезьян. Как у большинства видов, наследственная программа развития, записанная в молекулах ДНК с помощью универсального триплетного кода, организована у человека главным образом в его кариотипе. Хромосомный набор вида Homo sapiens состоит из 23 пар хромосом, содержащих 30—40 тыс. генов. Изучение структуры кариотипа и отдельных хромосом с использованием методик дифференциального (избирательного) окрашивания показало, что характер распределения красителя в разных хромосомах сходен у человека и человекообразных обезьян — шимпанзе и гориллы, имеющих 24 пары хромосом. Это сходство позволило сделать вывод, что одна из крупных хромосом человеческого кариотипа, очевидно, появилась в результате слияния двух акроцентрических хромосом обезьяноподобного предка (см. разд. 3.5.3.3). Изучение процессов мутагенеза обнаружило, что отдельные гены человека могут изменять свою структуру с частотой, соизмеримой с таковой у других живых организмов (10-5—10-6 на один ген на поколение). Правда, в силу социальности человек создает в ходе своей деятельности новую среду с более высокими дозами и более широким спектром мутагенов, что не может не сказываться на интенсивности мутационного процесса в наследственном материале не только человечества, но и других видов живых организмов. Таким образом, молекулярно-генетические механизмы обеспечения наследственности и изменчивости у человека имеют много общего с таковыми у других видов. Это делает возможным использование сведений, получаемых в ходе экспериментов на животных, для изучения наследственности и изменчивости у человека. 4.1. МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ МЕХАНИЗМЫ НАСЛЕДСТВЕННОСТИ И ИЗМЕНЧИВОСТИУ ЧЕЛОВЕКАБлагодаря большому объему человеческого генома и более низкому давлению естественного отбора, связанному с социальной природой человека (см. § 1,9), в генофонде человечества за тысячелетия его существования в результате постоянно идущего мутационного процесса накоплено большое число аллелей многих генов. Это является причиной формирования у людей разнообразных вариантов признаков и свойств как на структурном, так и на биохимическом уровнях. В основе индивидуальных различий по многим белкам лежат изменения соответствующих генов. Изучение аминокислотного состава вариантов белков человеческого организма, интенсивности их синтеза, функциональной активности дает ценные сведения об организации и экспрессии его наследственного материала. Удобной моделью для изучения молекулярно-генетических механизмов наследственности и изменчивости у человека является гемоглобин — специфический белок эритроцитов, легко выделяемый из организма без применения трудоемких биохимических методик. В результате длительного изучения этого белка накопилось много фактов, свидетельствующих об изменчивости его первичной структуры и свойств. В настоящее время обнаружено около 400 различных разновидностей гемоглобина, встречающихся как в нормальном развитии на разных стадиях онтогенеза, так и приводящих к различным заболеваниям. Молекула гемоглобина состоит из четырех полипептидных цепей (двух α- и двух β-цепей), каждая из которых соединена с небелковым компонентом — гемом, содержащим железо. Две названные полипептидные цепи имеют варианты, контролируемые разными, но близкими нуклеотидными последовательностями, которые образуют два семейства генов (см. разд. 3.6.4.3). Различные нуклеотидные последовательности экспрессируются на определенных стадиях индивидуального развития — у эмбриона, плода, после рождения (см. § 6.2). При этом полипептиды, сменяющиеся в зависимости от стадии онтогенеза, незначительно различаются по аминокислотному составу. Так, Aγ и Gγ-глобины различаются по одной аминокислоте в 136-м положении (аланин или глицин). Вариант Aγ-глобина (TAγ) в 75-м положении вместо изолейцина имеет треонин. Цепь δ отличается от β-цепи лишь десятью аминокислотными остатками. Из многочисленных мутаций гемоглобина большинство достаточно редки и лишь немногие из них встречаются чаще других, например HbS, HbC, НЬЕ. Большая часть вариантов гемоглобина (около 350) различается единичными аминокислотными заменами, причиной которых являются генные мутации, связанные с заменой оснований в нуклеотидных последовательностях α- или β-глобинового семейства. Многие аминокислотные замены существенно не влияют на функцию гемоглобина и не приводят к патологическим проявлениям. Как правило, это замены в обращенных наружу участках полипептидных цепей тетрамера. Замены аминокислот, нарушающие нормальную спиральную структуру цепей, часто вызывают неустойчивость гемоглобина. Замена в участках, которыми α- и β-цепи контактируют друг с другом, влияют на сродство гемоглобина к кислороду. Нарушения функций гемоглобина, возникающие в результате таких изменений структуры α- и β-глобиновых генов, ведут к появлению заболеваний, которые можно разделить на четыре основные группы. 1. Гемолитические анемии. Проявляются в распаде эритроцитов, зависящем от неустойчивости гемоглобина (описано около 100 вариантов нестабильных гемоглобинов с мутациями в гене β-цепи). 2. Метгемоглобинемии. Обусловлены ускоренным окислением двухвалентного железа до трехвалентного и образованием гемоглобина М (известны пять таких мутаций в генах α- и β-цепей, состоящих в замене одного основания). 3. Эритроцитоз. Заключается в образовании большего, чем обычно, количества эритроцитов, что обусловлено повышенным сродством гемоглобина к кислороду, который с трудом высвобождается в тканях (таких мутаций известно около 30). 4. Серповидно-клеточная анемия. Заключается в замене гемоглобина НЬА на HbS, который отличается растворимостью и кристаллизацией в условиях гипоксии, что приводит к изменению формы эритроцитов, и проявляется фенотипическим многообразием симптомов (см. рис. 3.21). Заболевания первых трех групп наследуются по доминантному типу, так что гетерозиготы по мутантному гену страдают нарушением здоровья. Наследование серповидно-клеточной анемии при обычных условиях осуществляется по рецессивному типу, но в условиях сильной гипоксии, например при нахождении на высоте свыше 3000 м над уровнем моря гетерозиготы НbА HbS также страдают анемией. Описанные мутантные формы гемоглобина возникают в результате изменений структуры генов по типу замены оснований. Мутации иного характера приводят к появлению аллелей глобинов, обусловливающих другие виды патологии. Так, нарушение процесса рекомбинации между аллельными генами (неравноценный кроссинговер) приводит к изменению числа нуклеотидов в них. Следствием этого может быть сдвиг рамки считывания. Нередким результатом таких структурных изменений генов является подавление синтеза той или иной цепи гемоглобина, приводящее к развитию патологических состояний, известных под общим названием талассемии. Деления одного нуклеотида в 139-м триплете α-глобинового гена, состоящего из 141 триплета, приводит к сдвигу рамки считывания и прочитыванию в новой рамке терминирующего 142-го кодона. При этом (α-глобиновая цепь удлиняется на пять дополнительных аминокислот. Такой особенностью α-цепей характеризуется гемоглобин Vayne. Когда деления располагается ближе к 5'-концу, активный продукт не синтезируется и развиваются различные формы α-, β- и γ-талассемий. Некоторые варианты гемоглобинов возникают в результате дупли-каций. Так, гемоглобин Grady несет дупликацию 116—118 аминокислотных остатков в γ-цепи. В гемоглобине Cranston удлинение р-цепи до 158 аминокислотных остатков является результатом дупликации AG-последовательности после 144-го триплета и последующего сдвига рамки с пропитыванием терминального кодона. Описанное выше свидетельствует о том, что различные отклонения в структуре ДНК глобиновых генов приводят к замене аминокислот или удлинению полипептидных цепей. Это является причиной образования многих вариантов гемоглобина, которые определяют развитие у человека заболеваний, наследующихся в ряду поколений. Не меньший интерес представляют механизмы развития различных заболеваний человека, в основе которых лежат мутации генов, приводящие к синтезу белков-ферментов со сниженной активностью или к его подавлению. Это нарушает течение процессов, катализируемых данными ферментами в клетках организма. Примером наследственно детерминированных повреждений метаболизма в организме человека служит фенилкетонурия, развивающаяся вследствие нарушения процессов обмена аминокислоты фенилаланина и накопления в организме токсических промежуточных продуктов. 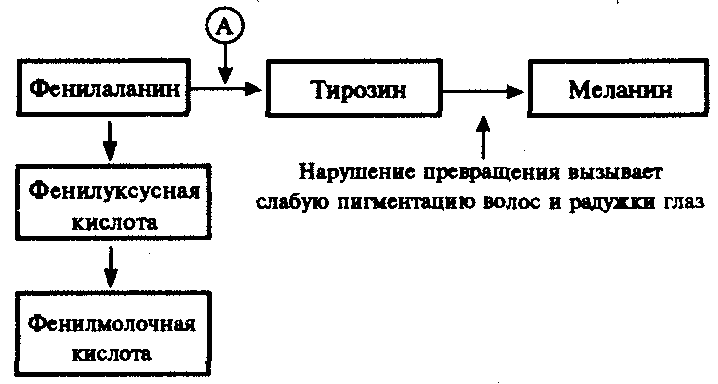 Рис. 4.1. Краткая схема обмена фенилаланина: А — фермент фенилаланингидроксилаза, наследственный дефект которого приводит к развитию фенилкетонурии При дефекте фермента фенилаланингидроксилазы фенилаланин не превращается в тирозин (рис. 4.1) и накапливается в крови больных в больших концентрациях (до 0,5—0,6 г/л вместо 0,003— 0,04 г/л в норме). Это приводит к частичному превращению фенилаланина в фенилуксусную и фенилмолочную кислоты, накопление которых наряду с повышенной концентрацией самого фенилаланина оказывает токсическое действие на мозг ребенка. В результате у детей наблюдается различная степень дефекта умственного развития. Нарушение метаболизма фенилаланина сопровождается также нарушением синтеза пигмента меланина, поэтому у больных наблюдается слабая пигментация волос и радужки глаз. Кроме того, высокая концентрация фенилаланина оказывает ингибирующее влияние на ряд ферментных систем, участвующих в превращении других аминокислот: у больных развивается судорожный синдром, нарастает отставание интеллектуального развития. Наследование фенилкетонурии осуществляется по рецессивному типу. Таким образом, рассмотренные выше примеры демонстрируют весь спектр действия молекулярно-генетических механизмов, обеспечивающих образование в человеческом организме белков как нормально функционирующих, так и обусловливающих развитие различных патологических состояний. Из сказанного по поводу гемоглобина следует, что, во-первых, образование главного функционального белка эритроцитов находится под генным контролем, во-вторых, формирование тетрамерной формы этого белка, с которой связана его физиологическая активность, требует взаимодействия неаллельных генов α- и β-глобинов. Специфический контроль небелковой части молекулы гемоглобина также имеет место и осуществляется независимо, через гены ферментов, необходимых для синтеза гема. Особенности проявления патологических признаков у носителей мутантных аллелей свидетельствуют о существовании определенных отношений между ними и нормальными аллелями. Так, аллель серповидно-клеточности в сочетании с нормальным аллелем (3-глобина (НbА HbS) проявляет себя в обычных условиях как рецессивный. Так же ведет себя мутантный аллель гена, детерминирующего синтез фермента фенилаланингидроксилазы. Проявлением взаимодействия между мутантным и нормальным аллелями по типу доминирования последнего является формирование в организме белка с нормальными свойствами у гетерозигот. Отсутствие нормального аллеля в генотипе организма, гомозиготного по мутантному аллелю, приводит к развитию патологического состояния, обусловленного нарушением функциональной активности соответствующего белка. Особую группу наследственно обусловленных патологических состояний у человека представляют заболевания, причиной которых являются мутации митохондриальной ДНК (мгДНК). Биосинтез митохондриальных белков находится под контролем двух генетических систем: ядерных и митохондриальных генов. Большая часть белков кодируется ядерной ДНК, синтезируется в цитоплазме, а затем транспортируется в митохондрии. Наряду с этим в кольцевой молекуле ДНК органеллы имеются гены, которые отвечают за собственный синтез белков, а также участвующих в нем тРНК и рРНК. В ядерном геноме имеется значительное количество генов, обеспечивающих функционирование митохондриальной ДНК. Предполагают, что мутации некоторых ядерных генов приводят к делениям значительных участков ДНК митохондрии. В результате нарушается синтез собственных белков, к числу которых относятся и ферменты дыхательных цепей, нарушается дыхательная функция митохондрии. У человека описано более 100 заболеваний, причиной которых являются изменения в структуре мтДНК (см. 6.4.1.4). 4.2. КЛЕТОЧНЫЕ МЕХАНИЗМЫ ОБЕСПЕЧЕНИЯ НАСЛЕДСТВЕННОСТИ И ИЗМЕНЧИВОСТИУ ЧЕЛОВЕКАВ генетическом материале человека в ряде ситуаций возникают изменения, которые, непосредственно не затрагивая отдельных генов, вызывают серьезные нарушения в состоянии организма. Такие изменения чаще всего касаются структуры хромосом или их числа в клетках. Результатом этого является нарушение баланса генов, т.е. того соотношения доз различных аллелей, которое требуется для нормального развития признаков и организма в целом. Постоянство кариотипа поддерживается в ряду клеточных поколений благодаря митозу. В ряду поколений организмов это постоянство обеспечивается сочетанием мейоза и оплодотворения. Нарушение митоза и мейоза, обусловливающих закономерное распределение хромосом при образовании соматических и половых клеток, может служить причиной изменения строения и числа этих ядерных структур. Нередко хромосомные перестройки появляются в результате воздействия на клетки внешних факторов. К таким факторам относится, например, ионизирующее излучение, вызывающее разрывы хромосом и последующие изменения их структуры. У человека описаны также случаи наследственно обусловленной неустойчивости хромосом, их сверхчувствительности к действию агентов различной природы, приводящих к хромосомным разрывам. Это наблюдается при анемии Фанкони, синдроме Блума, атаксии-телеангиэктазии, пигментной ксеродерме. Так, при пигментной ксеродерме высокая чувствительность к ультрафиолетовому свету, сопровождающаяся повышенной ломкостью хромосом, связана с наследственно обусловленным нарушением репарации ДНК. Изменение числа хромосом, как правило, является результатом нарушения нормального течения клеточных делений, что приводит к образованию анэуплоидных и полиплоидных соматических клеток или гамет с аномальным числом хромосом. Повреждения механизмов обеспечения наследственности, действующих на клеточном уровне, в масштабе организма приводят к разным результатам. Так, мутации в соматических клетках организма (соматические мутации) могут приводить к различным заболеваниям особи, однако без передачи их потомству при половом размножении. Нарушения наследственной программы в половых клетках (генеративные мутации), не Проявляясь в фенотипе данного организма, ведут к появлению мутантного потомства. Следовательно, точное воспроизведение определенных наследственных характеристик в ряду поколений клеток организма способствует поддержанию здоровья данной особи. Залогом появления здорового в наследственном отношении потомства является в первую очередь сбалансированность генома родительских гамет, содержащего благоприятные аллели генов. При наличии в геноме гаметы одного из родителей «неблагоприятных» аллелей генов их действие может снижаться в результате взаимодействия с нормальными аллелями другого родителя. 4.2.1. Соматические мутацииМутации различного ранга (генные, хромосомные или геномные), возникающие в соматических клетках организма, наследуются потомками этих клеток и делают организм мозаиком, т.е. особью со смешанными популяциями клеток. В разд. 3.6.5.1 и 3.6.5.2 рассмотрены примеры естественного мозаицизма женского организма по активно функционирующим в его клетках Х-хромосомам и связанное с этим явление аллельного исключения, когда в разных клетках организма экспрессируются разные аллели Х-сцепленных генов. К примеру, у женщины — гетерозиготной носительницы рецессивного аллеля гемофилии — степень нарушения свертывающей системы крови зависит от соотношения соответствующих клеток с генетически инактивированными Х-хромосомами, несущими нормальный или му-тантный аллель. Нередко у человека встречается мозаицизм по геномным мутациям, связанный с нарушением расхождения хромосом при митозе. Например, в случае синдрома Дауна (трисомия по 21-й хромосоме) мозаицизм встречается с частотой 2 на 48 пациентов, а в популяции их частота равна 1 на 31 000. Чем раньше в ходе развития организма происходит нарушение деления соматических клеток, сопровождающееся нерасхождением дочерних хромосом к полюсам ахроматинового веретена, тем более выраженной будет симптоматика заболевания, вызываемого данной анэуплоидией. Нарушение митоза на более поздних стадиях индивидуального развития приводит к локальному мозаицизму, который может не сопровождаться выраженными отклонениями от нормы. В этом случае наиболее опасным является мозаицизм клеток генеративных тканей, из которых с достаточно большой вероятностью организм может образовывать гаметы с аномальным числом хромосом. Иногда возникающие соматические мутации являются причиной появления злокачественных новообразований. На рис. 4.2 представлена последовательность событий, приводящих к образованию таких опухолей. Как правило, в основе лежит повреждение ДНК, вызываемое внутренними факторами (нарушением процессов репликации, репарации или рекомбинации) или внешними воздействиями (ионизирующей радиацией, химическими мутагенами или вирусами). Одним из результатов такого повреждения ДНК может оказаться появление клона клеток, обладающего дефектами регуляции клеточного размножения, что приводит к опухолевому росту. Причиной злокачественного разрастания ткани могут быть также нарушение митоза и неравноценное распределение хромосом между дочерними клетками с возникновением анэуплоидий или хромосомных аберраций. Это вызывает либо гибель клеток, либо приводит к появлению клонов, способных к неконтролируемому росту. В злокачественных образованиях обычно встречаются субклоны, имеющие разные кариотипы, что свидетельствует о множественных аномалиях митоза в клетках опухолей. 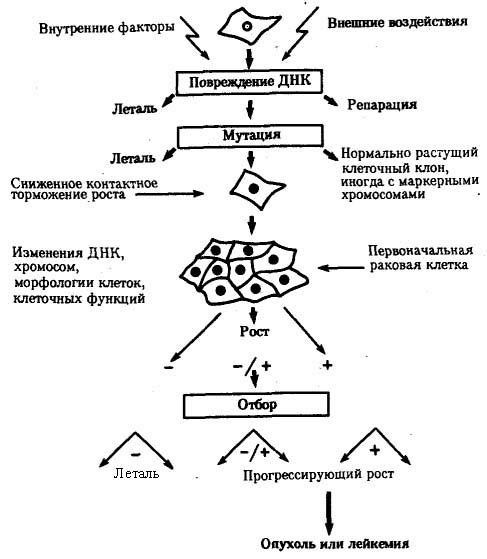 Рис. 4.2. Соматические мутации как причина злокачественного перерождения тканей Так как в основе злокачественного перерождения тканей может лежать изменение наследственного материала клеток, становится очевидной важная роль мутагенных факторов в процессе возникновения опухолей. Одним из таких мутагенных факторов являются вирусы, которые могут индуцировать в хромосомах то или иное мутационное изменение. Среди опухолей человека вирусное происхождение имеет лимфома Беркитта. Пониманию молекулярно-биологических механизмов опухолевого роста способствовало открытие так называемых онкогенов, вызывающих злокачественное перерождение клеток и входящих в состав генома ретровирусов. Геном ретровируса состоит из одноцепочечной РНК и содержит ген обратной транскриптазы. При проникновении вируса в клетку-хозяина под контролем фермента обратной транскриптазы образуются множественные копии генетической информации вируса, но уже в виде двухцепочечной ДНК, которые встраиваются в ДНК клетки-хозяина. Функционирование таких ДНК-копий в составе генома клетки приводит к синтезу вирусных РНК и белков, причем вирусный онкоген (v-onc) трансформирует клетку-хозяина в опухолевую. Использование ДНК-зондов на основе ретровирусных онкогенов обнаружило их гомологию некоторым собственным нуклеотидным последовательностям генома клеток. Эти участки получили название протоонкогенов или клеточных онкогенов (c-onc). Протоонкогены участвуют в контроле клеточного роста, но в обычном состоянии не приводят к опухолевой трансформации. Их мутантные аллели, стимулируя митоз, могут вызвать рост опухолей. Иногда активация клеточного онкогена обусловливается единичной точковой мутацией в нем. В других случаях нет необходимости в такой мутации, так как трансформирующий эффект наблюдается при присоединении онкогена к фрагментам ДНК, обладающим сильными промоторными свойствами. В таких ситуациях следует допустить, что протоонкогенам присущи свойства транспозонов, или «прыгающих генов». Предполагают, что вирусные онкогены на каком-то этапе эволюции произошли от клеточных онкогенов, интегрированных в геном вируса. Причины, обусловливающие большую трансформирующую активность вирусных онкогенов в сравнении с клеточными, до настоящего времени не вполне ясны. В опухолевых клетках часто наблюдаются хромосомные аномалии, причем некоторые опухоли отличаются наличием специфических хромосомных дефектов. Установлено, что онкогены нередко обнаруживаются в непосредственной близости от точек разрывов, происходящих при опухолеспецифичных хромосомных перестройках. Это подтверждает их роль в злокачественной трансформации клеток. 4.2.2. Генеративные мутацииИзменения наследственной программы половых клеток человека приводят к рождению потомства с различными наследственно обусловленными болезнями, в зависимости от ранга мутаций — генными или хромосомными. Различные генные мутации по-разному сказываются на жизнеспособности организма, причем в случае их рецессивности они могут долго не проявляться фенотипически у потомков. Хромосомные перестройки и геномные мутации приводят к выраженным отклонениям в развитии и часто являются причиной гибели организма на разных стадиях его онтогенеза, обычно в раннем эмбриогенезе. В значительной степени именно этими мутациями определяется высокий процент (15%) прерывания диагностированных беременностей. Триплоидии плода, как правило, приводят к прерыванию беременности на ранних стадиях, однако описано очень небольшое число случаев живорождения триплоидов. Анэуплоидия по разным хромосомам встречается как в материале абортусов, так и у рожденных детей. Некоторые анэуплоидий несовместимы с жизнью. Так, трисомия по 16-й хромосоме обнаруживается только в материале абортусов. В то же время у человека известны синдромы, связанные с аномалиями числа хромосом, характеризующиеся разной степенью жизнеспособности.  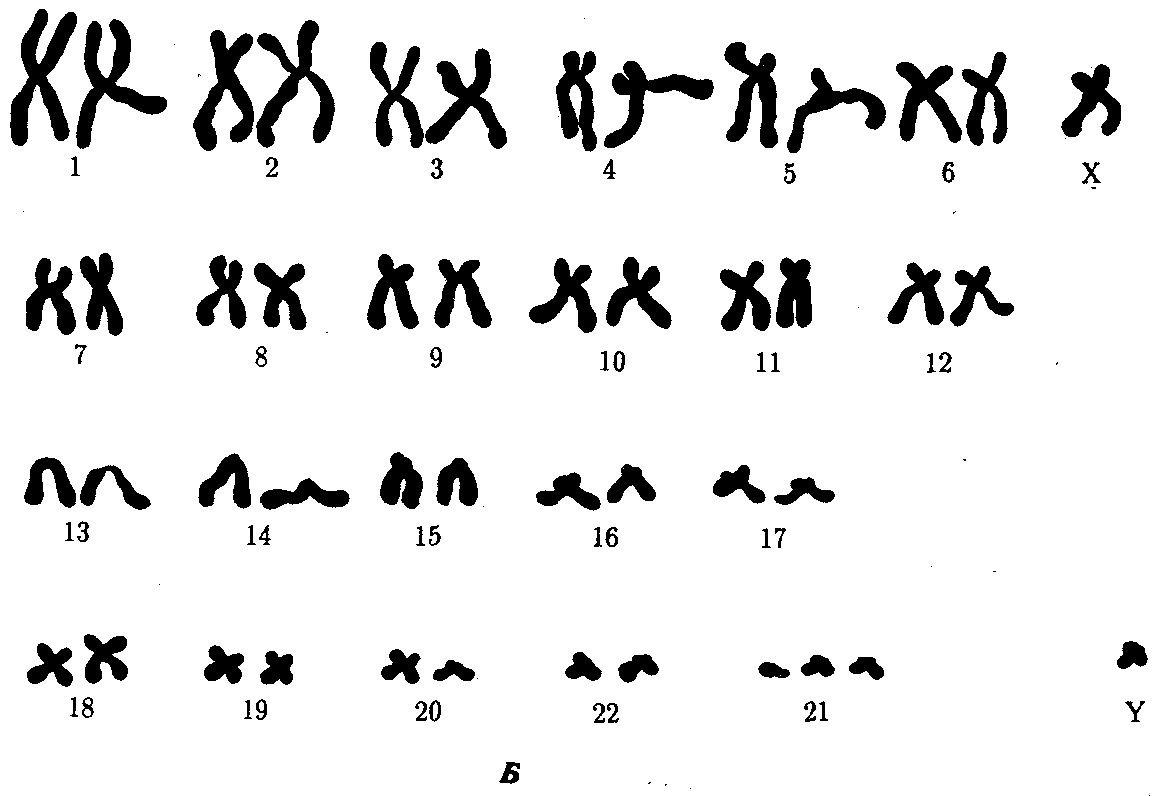 Рис. 4.3. Синдром трисомии 21 (синдром Дауна). А — внешний вид больного; Б —кариотип больного Наиболее частым хромосомным заболеванием у человека является синдром Дауна, обусловленный три-сомией по 21-й хромосоме, встречающийся с частотой 1—2 на 1000 (рис. 4.3). Примерно в 60% случаев трисомия 21 является причиной гибели плода, около 30% родившихся умирает на первом году жизни. Еще 46% не переживает Злетний рубеж, однако иногда люди с синдромом Дауна доживают до значительного возраста (рис. 4.4), хотя в целом продолжительность их жизни сокращена. Применение эффективных противомикробных препаратов позволяет несколько увеличить продолжительность жизни таких больных. Трисомия 21 может быть результатом случайного нерасхождения гомологичных хромосом в мейозе. Наряду с этим известны случаи регулярной трисомии, связанной с транслокацией 21-й хромосомы на другую —21, 22, 13, 14 или 15-ю хромосому (рис. 4.5).  Рис. 4.4. Женщина с синдромом Дауна в возрасте 38 лет 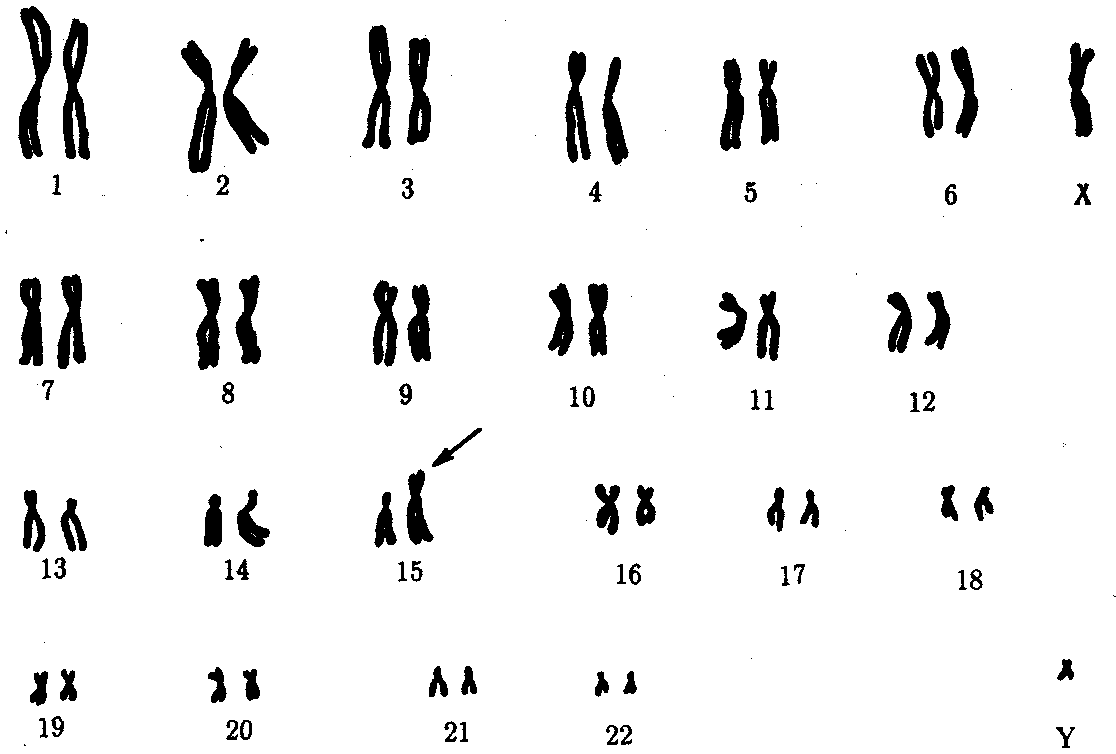 Рис. 4.5. Кариотип при транслокационном синдроме Дауна (одна 21-я хромосома присоединена к 15-й хромосоме — указано стрелкой) 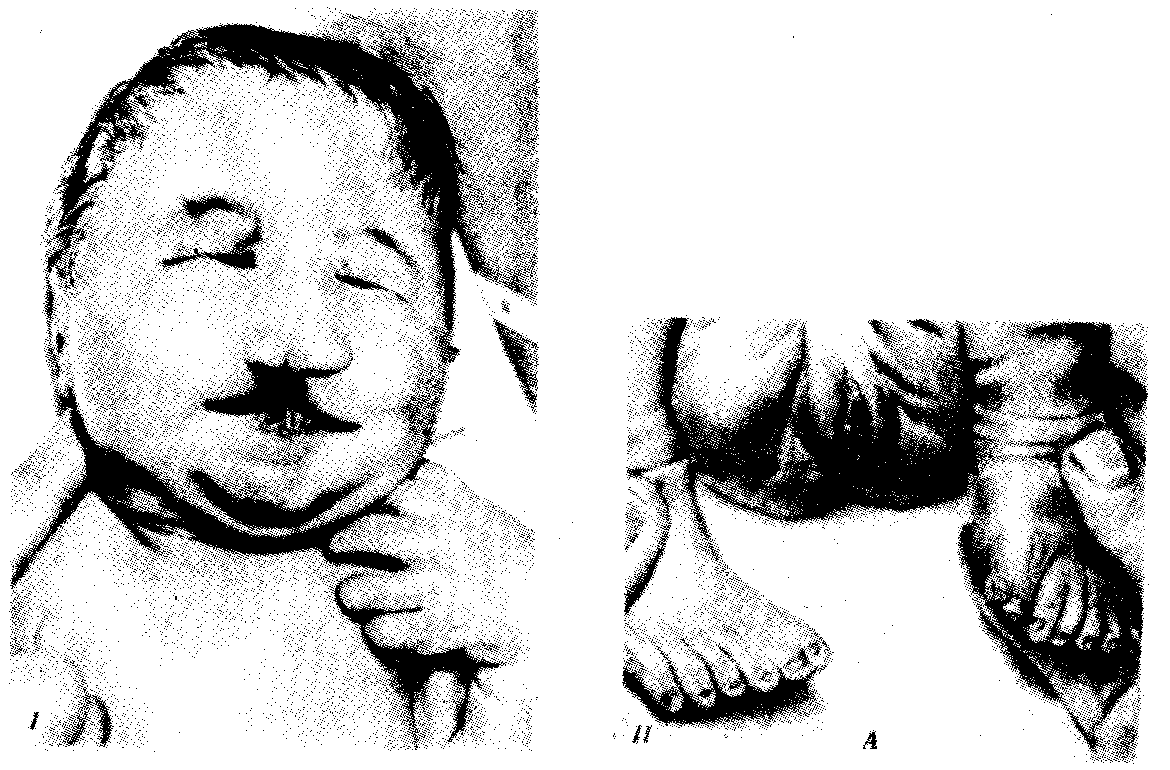 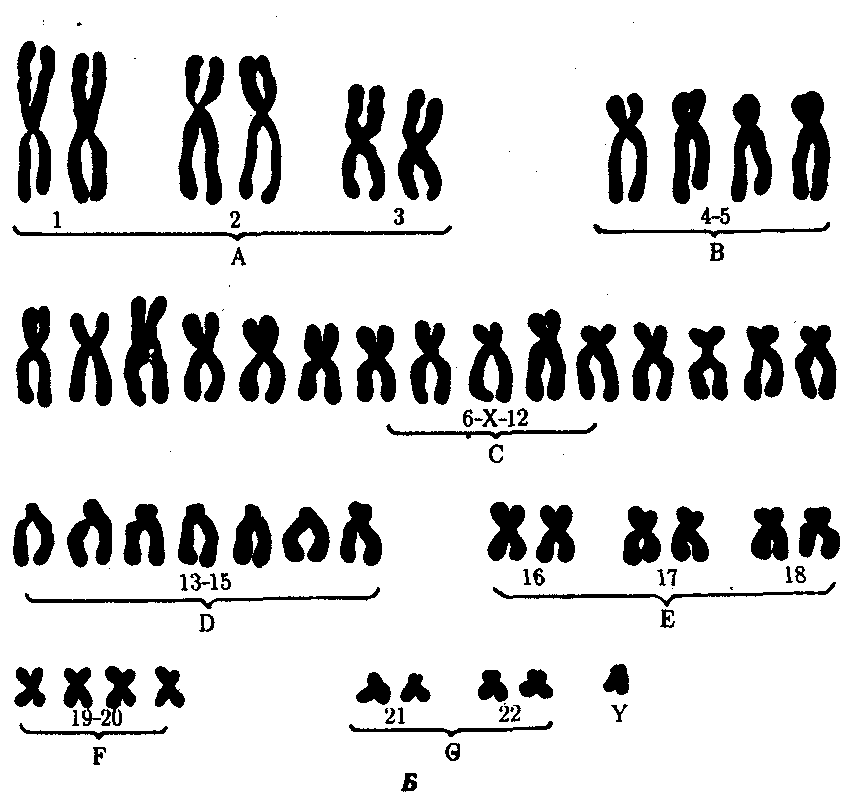 Рис. 4.6. Синдром трисомии 13 (синдром Патау). А — внешний вид больного; Б —кариотип больного с трисомией в группе D: I — аномалии лица, II — двусторонняя полисиндактилия стоп  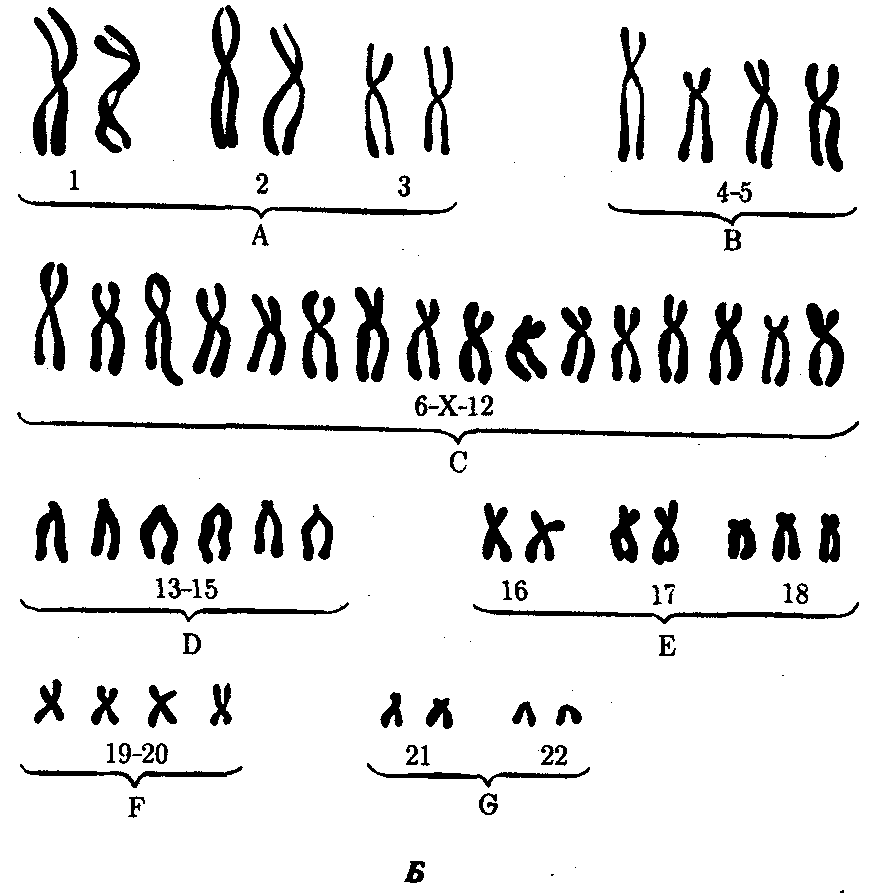 Рис. 4.7. Синдром трисомии 18 (синдром Эдвардса). А — внешний вид больного; Б — кариотип больного при трисомии в группе Е: I — черепно-лицевые аномалии, II — характерное расположение пальцев на кистях больного  Среди других аутосомных трисомий известны трисомии по 13-й хромосоме — Синдром Патау (рис. 4.6), а также по 18-й хромосоме — синдром Эдвардса (рис. 4.7), при которых жизнеспособность новорожденных резко снижена. Они гибнут в первые месяцы жизни из-за множественных пороков развития. Применение методов дифференциального окрашивания хромосом позволило открыть три новых синдрома, обусловленных трисомиями по 8, 9 и 22-й хромосомам, при которых также наблюдаются тяжелые комплексные пороки развития (рис. 4.8). Достаточно часто у человека встречаются анэуплоидии по половым хромосомам (рис. 4.9—4.11). В отличие от анэуплоидии по аутосомам дефекты умственного развития у больных выражены не столь отчетливо, у многих оно в пределах нормы, а иногда даже выше среднего. Вместе с тем у них постоянно наблюдаются нарушения развития половых органов и гормонозависимого роста тела. Реже встречаются пороки развития других систем. Относительно благоприятные последствия увеличения числа Х-хромосом, видимо, связаны с возможностью компенсации дозы соответствующих генов благодаря естественной генетической инактивации этих хромосом, а также мозаичному характеру такой инактивации. Среди анэуплоидных синдромов по половым хромосомам моносомия Х (ХО) (синдром Шерешевского — Тернера) встречается много реже, чем трисомия X, синдром Клайнфельтера (XXY, XXXY), а также XYY, что указывает на наличие сильного отбора против гамет, не содержащих половых хромосом, или против зигот ХО. Это предположение подтверждается достаточно часто наблюдаемой моносомией Х среди спонтанно абортированных зародышей. В связи с этим допускается, что выжившие зиготы ХО являются результатом не мейотического, а митотического нерасхождения, или утраты Х-хромосомы на ранних стадиях развития (см. рис. 4.9). Моносомии YO у человека не обнаружено.  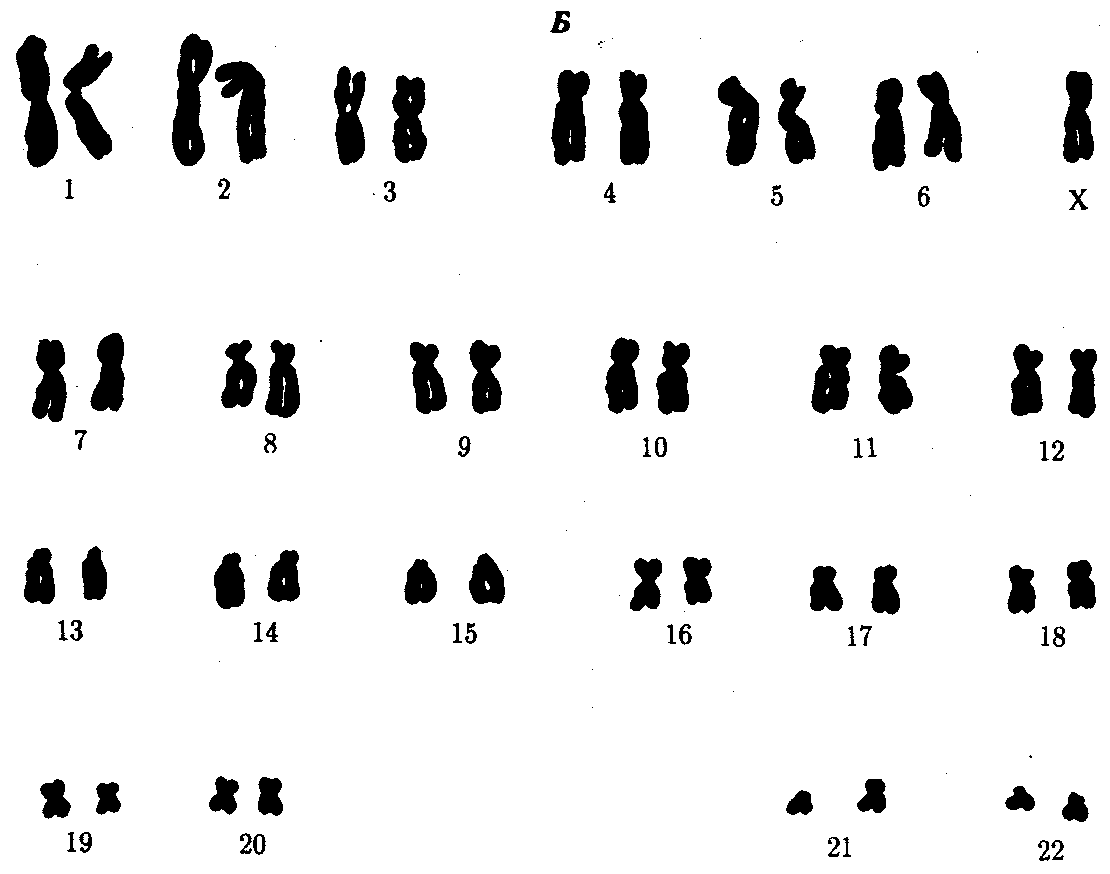 Рис. 4.9. Синдром моносомии Х (ХО-синдром, синдром Шерешерского — Тернера). А — внешний вид больной; Б — кариотип женщины с синдромом ХО: I — выраженная трапециевидная шейная складка, широкая грудная клетка, широко расставленные, слаборазвитые соски молочных желез, II — характерные лимфатические отеки на ногах 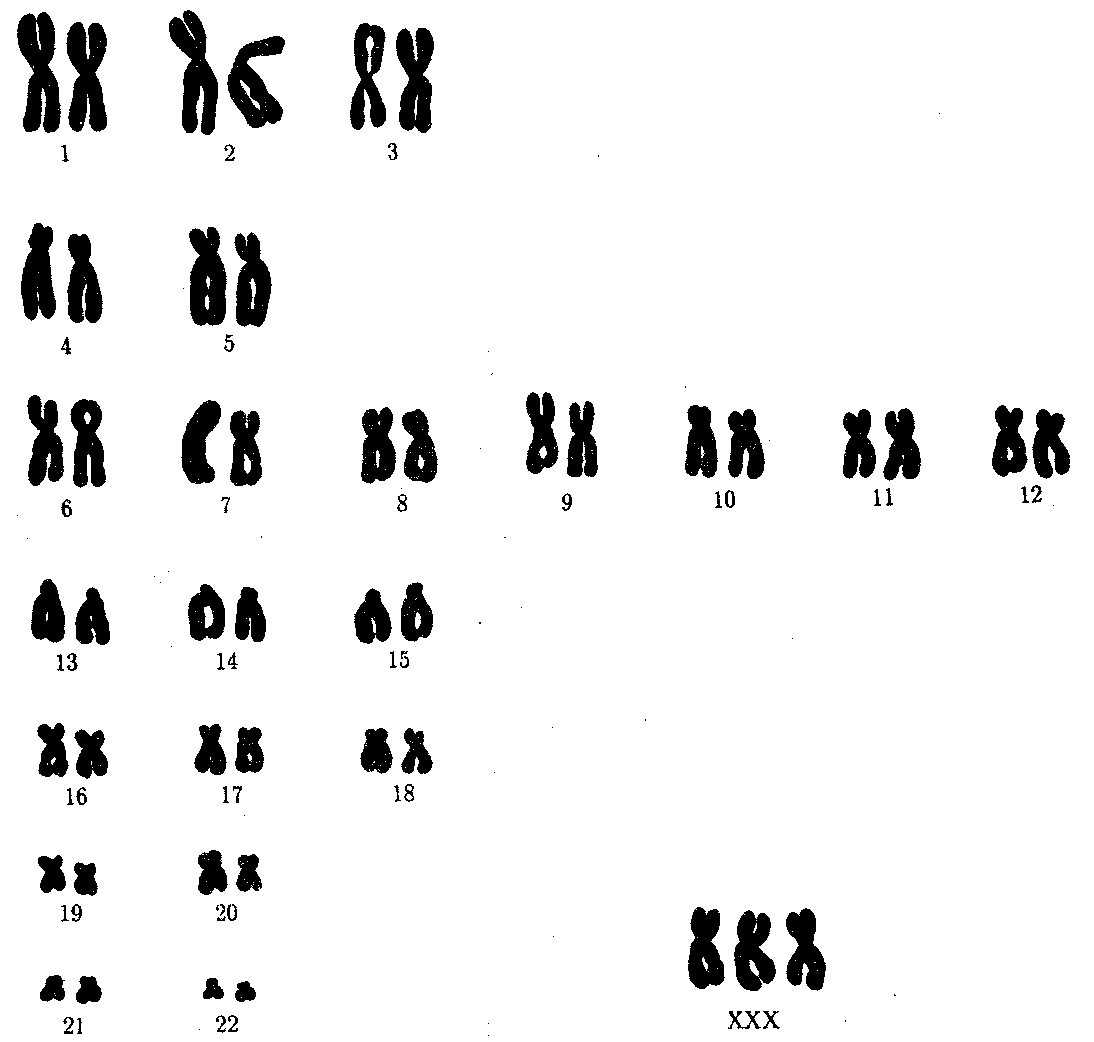 Рис. 4.10. Кариотип женщины с синдромом трисомии Х 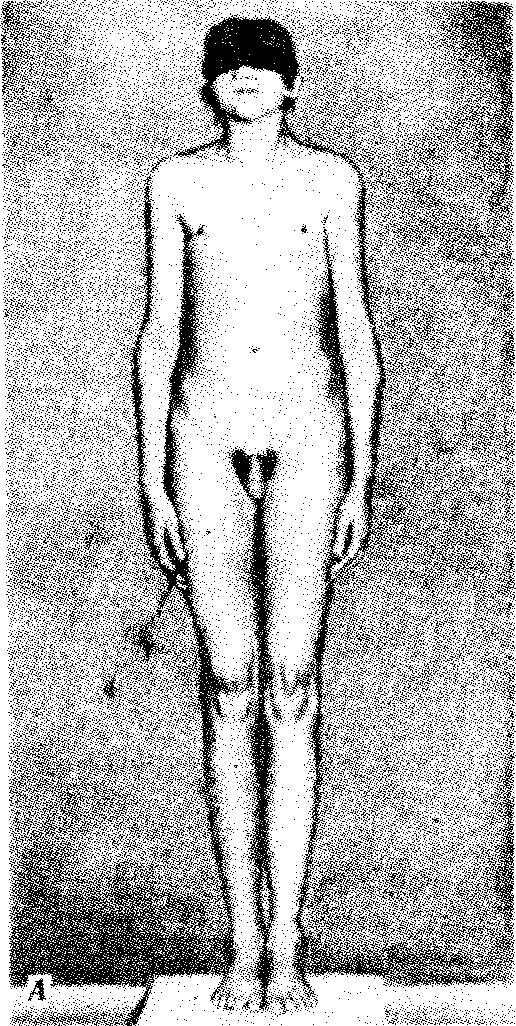 Рис. 4.11. Синдром Клайнфельтера. А —внешний вид больного (характерен высокий рост, непропорционально длинные конечности); Б—кариотип больного (XXY) Организмы с анэуплоидией по половым хромосомам при наличии Y-хромосомы развиваются по мужскому типу и фенотипически дают синдром Клайнфельтера (рис. 4.11). Это является еще одним свидетельством в пользу расположения фактора, определяющего мужской тип развития в Y-хромосоме. Из синдромов, связанных со структурными аномалиями хромосом, известен транслокационный синдром Дауна (см. рис. 4.5), при котором число хромосом в кариотипе формально не изменено и равно 46, так как дополнительная 21-я хромосома транслоцирована на одну из акроцентрических хромосом. При транслокации длинного плеча 22-й хромосомы на 9-ю развивается хронический миелолейкоз. При делении короткого плеча 5-й хромосомы развивается синдром кошачьего крика, при котором наблюдаются общее отставание в развитии, низкая масса при рождении, лунообразное лицо с широко расставленными глазами и характерный плач ребенка, напоминающий кошачье мяукание, причиной которого является недоразвитие гортани (рис. 4.12). 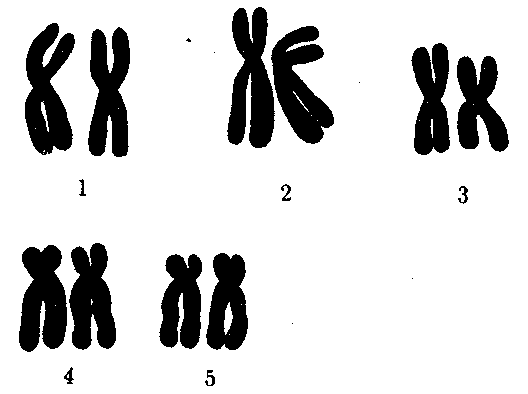 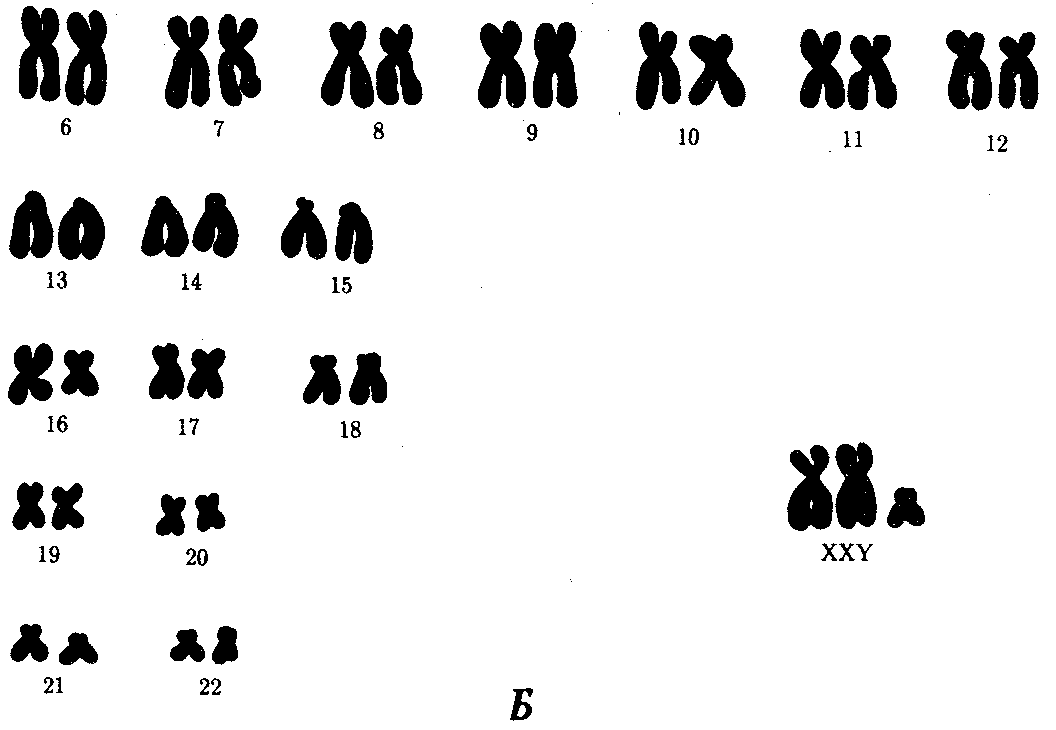   Рис. 4.12. Синдром хромосомы 5р (синдром кошачьего крика). А — внешний вид больного; Б — деления короткого плеча 5-й хромосы У носителей некоторых перицентрических инверсий нередко наблюдаются аномалии в виде умственной отсталости той или иной степени и пороков развития. Довольно часто такие перестройки наблюдаются в 9-й хромосоме человека, однако они существенно не влияют на развитие организма. Таким образом, нарушение наследственной программы организма, развивающегося из аномальных гамет, или мозаицизм его клеток, связанный с соматическими мутациями, являются причиной либо гибели организма, либо более или менее выраженного снижения его жизнеспособности. |