Лекция 09 (Патологии нервной системы). Лекции Цель
 Скачать 0.54 Mb. Скачать 0.54 Mb.
|

Комплекс внутриклеточных процессов, возникающих при ишемии и вызывающих дегенерацию и гибель мембранВесьма чувствительны к аноксии тормозные механизмы. Одним из следствий этого является растормаживание неповрежденных структур ЦНС. На ранних стадиях ишемии, когда нейроны мозга еще способны давать реакцию, они могут гиперактивироваться. На поздних стадиях ишемии гиперактивация нейронов сменяется их инактивацией. С поступлением Na+ в нейрон связана первая, острая фаза поражения нейрона. Возрастание концентрации Na+ в цитозоле нейрона приводит к повышению осмолярности, что обусловливает вход воды в нейрон и его набухание. В дальнейшем повышение осмолярности нейрона связано также с накоплением в нем Са2+, молочной кислоты, неорганического фосфора. С входом Са2+ в нейрон связана вторая фаза повреждения нейрона. Увеличение количества Са2+, поступающего в нейрон, обусловливается активацией глутаматных рецепторов в связи с усиленным выделением глутамата нервными окончаниями при ишемии. Антагонисты глутаматных рецепторов и антагонисты Са2+ (блокаторы Са2+-каналов) способны предотвратить ишемическую дегенерацию нейронов и оказать лечебный эффект. Повреждение нейрона происходит не только во время ишемии, но и в связи с реперфузией мозга и возобновлением циркуляции крови. Именно они могут представлять главную опасность. Большую роль в реперфузионных постишемических повреждениях играют: новая волна поступления Са2+ в нейрон, ПОЛ (перекисное окисление липидов) и процессы свободнорадикального окисления, усиленные в связи с действием поступающего кислорода. Увеличивается содержание молочной кислоты из-за поступления глюкозы в условиях нарушения окислительного фосфорилирования и возросшего анаэробного гликолиза. Происходит отек мозга за счет поступления воды из крови при возобновлении циркуляции.
В сложный комплекс Са2+-индуцируемых внутриклеточных повреждений входят: альтерация внутриклеточных белков, усиленный фосфолипазный гидролиз и протеолиз, разрушение внутриклеточных структур, повреждение цитоплазматической и внутриклеточных мембран, набухание нейронов, нарушение деятельности генома. При критическом возрастании интенсивности этих процессов происходят необратимые повреждения и гибель нейрона, возникает так называемая кальциевая смерть. На поздних стадиях патологического процесса, вызванного ишемией мозга, а также при хронизации процесса возникает новый комплекс вторичных изменений - дегенеративно-дистрофические процессы, нарушения энзимных и метаболических систем, сосудистые изменения, образование антител к мозговой ткани, аутоиммунная агрессия и др. Они составляют патогенетическую структуру постишемической энцефалопатии, которая может продолжать развиваться (прогредиентное развитие). Эти процессы, а также изменения в других системах и органах с их последствиями имеют место и после реанимации организма, особенно если она была затяжной и поздней. В своей совокупности они составляют патогенетическую структуру постреанимационной болезни (В.А. Неговский). Гипоксия той или иной степени сопровождает многие (если не все) формы патологии мозга. Являясь типовым и неспецифическим процессом, она, однако, может вносить значительный вклад в его развитие. Вместе с тем умеренная гипоксия может стимулировать метаболические и пластические процессы в нейроне, способствовать адаптации и повышению резистентности, повышать трофический и пластический потенциал нейрона, усиливать адаптационные возможности мозга.
Синаптическая стимуляция и повреждение нейронов Возбуждающая синаптическая стимуляция может играть важную роль в развитии патологии нейрона. Усиленная и длительная синаптическая стимуляция сама по себе вызывает функциональное перенапряжение нейрона, которое может завершиться дегенерацией внутриклеточных структур. Эти повреждения усиливаются при нарушениях микроциркуляции и мозгового кровообращения, действии токсических факторов. Первостепенное значение синаптическая стимуляция имеет при развитии аноксических (ишемических) повреждений. Культура тканей нейронов становится чувствительной к аноксии лишь после установления синаптических контактов между нейронами. Синаптическая стимуляция реализуется через действие возбуждающих аминокислот (глутамат, аспартат, L-гомоцистеинат), причем эти повреждения подобны тем, которые возникают при ишемии и связаны с увеличенным содержанием внутриклеточного Са2+. Этот эффект известен как нейротоксическое (или цитотоксическое) действие возбуждающих аминокислот. С синаптической гиперактивацией, действием возбуждающих аминокислот и гипоксией связаны повреждение и гибель нейронов при эпилептическом статусе и в постишемическом периоде. При этом к патогенному действию указанных факторов присоединяется энергетический дефицит. В связи с изложенным становятся понятными благоприятные эффекты (т.е. ослабление синаптического воздействия) уменьшения функциональной нагрузки, предотвращение дополнительных раздражений, «охранительное», по И.П. Павлову, торможение обратимо поврежденных нейронов. Нарушение деятельности нейрона при изменении процессов внутриклеточной сигнализации После восприятия рецептором сигнала (связывания рецептором нейромедиатора, гормона и др.) в нейроне возникает каскад цепных метаболических процессов, обеспечивающих необходимую активность нейрона. Важную роль в этих процессах играют так называемые усилительные, или пусковые, ферменты и образующиеся под их влиянием вещества-посредники, вторичные мессенджеры .
Совокупность каскадных мембранных и внутриклеточных процессов составляет эндогенную усилительную систему нейрона, которая может обеспечить многократное усиление входного сигнала и возрастание его эффекта на выходе из нейрона. Так, каскад метаболических процессов аденилатциклазного пути может усилить эффект стимула в 107-108 раз. Благодаря этому возможны выявление и реализация слабого сигнала, что имеет особое значение в условиях патологии, при нарушении синаптического проведения. Многие изменения функций нейрона связаны с действием патогенных агентов на определенные звенья систем внутриклеточной сигнализации. Фармакологическая коррекция деятельности нейрона и эффекты лечебных средств также реализуются через соответствующие изменения этих систем. Так, холерный и коклюшный токсины действуют на процессы, связанные с активностью мембранных G-белков, активирующих или угнетающих аденилатциклазу. Ксантины (теофиллин, кофеин) обусловливают накопление цАМФ, что приводит к усилению деятельности нейрона. При действии ряда противосудорожных препаратов (например, дифенилгидантоина, карбамазепина, бензодиазепинов) и психотропных средств (например, трифтазина) угнетаются разные пути фосфорилирования белков, благодаря чему снижается активность нейронов. Ионы лития, применяемые при лечении некоторых эндогенных психозов, ослабляют деятельность системы фосфоинозитидов. С усиленным входом Са2+ связана эпилептизация нейронов, блокада этого входа антагонистами Са2+ подавляет эпилептическую активность. Гиперактивность нейрона Гиперактивность нейрона обусловлена значительным, выходящим из-под контроля нарушением баланса между возбуждением и торможением нейрона в пользу возбуждения. В функциональном отношении она заключается в продуцировании нейроном усиленного потока импульсов, который может иметь различный характер: высокочастотные потенциалы действия; отдельные разряды; разряды, сгруппированные в пачки, и пр. Особый вид гиперактивности представляет собой пароксизмальный деполяризационный сдвиг (ПДС) в мембране, на высоте которого возникает высокочастотный разряд .Такой вид гиперактивности рассматривается как проявление эпилептизации нейрона.
Указанный сдвиг баланса между возбуждением и торможением может быть обусловлен либо первичным усиленным возбуждением нейрона, преодолевающим тормозной контроль, либо первичной недостаточностью тормозного контроля. Первый механизм реализуется значительной деполяризацией мембраны и усиленным входом Na+ и Са2+ в нейрон, второй - расстройством механизмов, обеспечивающих гиперполяризацию мембраны: нарушением выхода К+ из нейрона и входа Cl- в нейрон. Существенным эндогенным регулятором активности нейрона является γ-аминомасляная кислота (ГАМК). Она вызывает торможение нейрона при связывании со своим рецептором. В результате усиливается поступление Cl- в нейрон. 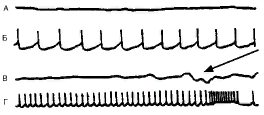 Различные виды спонтанной активности нейрона в эпилептическом очаге, вызванном столбнячным токсином в двигательной зоне коры головного мозга кошки. Кривые А и В - потенциалы, регистрируемые с поверхности мозга в эпилептическом очаге (ЭкоГ). Кривые Б и Г - запись электрической активности нейронов, выполненные с помощью внутриклеточного отведения. Нейрон может генерировать с разной частотой регулярные потенциалы действия. На кривой Г показано завершение потенциала высокочастотными разрядами. В это время на ЭкоГ (кривая В) в зоне очага появляется спайковый разряд (указан стрелкой) При растормаживании нейрона в связи с ослаблением торможения и деполяризацией мембраны происходит усиление поступления Са2+ в нейрон. Кроме того, Са2+, находясь уже в цитозоле, нарушает поступление С1- в нейрон, ослабляя, таким образом, изнутри ГАМКергическое торможение. С этим связана эпилептизация нейрона, возникающая под влиянием конвульсантов, которые нарушают ГАМКергическое торможение. Многие конвульсанты (например, пенициллин, коразол и др.) оказывают сложное действие на нейрон, одновременно активируя возбуждающие и инактивируя тормозные механизмы.
Хроническая стимуляция нейрона (например, при прямом электрическом раздражении, синаптическом воздействии, под влиянием возбуждающих аминокислот и др.) даже слабой интенсивности может с течением времени привести к гиперактивации нейрона. С другой стороны, выключение афферентации нейрона также обусловливает его гиперактивацию. Этот эффект объясняется повышением чувствительности нейрона и нарушением тормозных процессов. Таким образом, патологическая гиперактивация нейронов, их эпилептизация представляет сложный комплекс разнообразных мембранных и внутриклеточных процессов. Для подавления эпилептической активности целесообразно комплексное применение веществ, нормализующих основные патогенетические звенья процесса. Среди корригирующих воздействий первостепенное значение имеют блокада поступления Са2+ и восстановление тормозного контроля. |