Основы фармацевтического качества. Лекции Понятие и особенности термина качество лекарственного средства
 Скачать 1.33 Mb. Скачать 1.33 Mb.
|

|
«Основы фармацевтического качества» Цель лекции: Сформировать представление о качестве, жизненном цикле лекарственного средства и его связи с системой качества в глобальном масштабе. Обобщить представление о надлежащих практиках в фармации. План лекции: 1. Понятие и особенности термина «качество лекарственного средства» 2. Особенности фармацевтической продукции. 3. Понятие о надлежащих практиках в фармации (GХР). 1. Понятие и особенности термина «качество лекарственного средства» Известны многочисленные определения термина «качество». Например, одно из наиболее удачных звучит следующим образом: «Качество - это совокупность характеристик объекта, относящихся к его особенности удовлетворять установленные и предполагаемые потребности». В отношении лекарственных средств (ЛС) установленные и предполагаемые потребности - это необходимость диагностики, профилактики, лечения, изменения состояния и функций организма и пр., т. е. то, что ожидают получить от лекарственного средства его потребители - пациенты и врачи. Таким образом, в широком смысле слова термин КАЧЕСТВО в отношении лекарственного средства будет означать его способность удовлетворять существующие и предполагаемые потребности при медицинском либо ветеринарном применении. 2. Особенности фармацевтической продукции Фармацевтическая продукция как товар принципиально отличается от обычной продукции массового потребления. Наиболее характерные отличия можно продемонстрировать на примере использования рецептурных лекарственных препаратов. Основные отличия: 1) Потребитель не сам принимает решение о покупке лекарственного средства (по крайней мере, в отношении наиболее важных в терапевтическом или профилактическом отношении рецептурных препаратов). 2) Ни врач, принимающий решение о покупке лекарственного средства, ни сам покупатель не могут оценить качество в широком смысле слова, т. е. потребительские свойства предлагаемых к продаже препаратов. 3) Врач, принимающий решение о покупке лекарства, не оплачивает его. 4) При повышении цен на лекарственное средство спрос на него снижается слабо. 5) Существует риск в различии характеристик ЛС на этапе доказательства его эффективности и относительной безопасности при доклинических и клинических исследованиях и того же самого средства, но производимого в промышленных масштабах. 6) Приемлемость серийной продукции проверяется по показателям качества, входящих в спецификацию, т. е. по косвенным, техническим характеристикам. Однако они не всегда связаны с потребительскими свойствами лекарственного средства (например, скоростью и степенью всасываемости в системный кровоток, от чего напрямую зависит эффективность дозировки). 7) Купив лекарственный препарат и убедившись, что он ему не подходит, пациент не может вернуть ни препарат, ни деньги, ни здоровье. Обнаружив при использовании лекарства дефект, потребитель не может (за редкими исключениями) поменять его на другой, бездефектный. 8) Поскольку основной вид проверки качества лекарственных препаратов - разрушающий, практически невозможно применять полный 100% контроль с удалением бракованных единиц продукции. Именно по этой причине используются эффективные системы обеспечения качества и выборочный контроль. 9) В общегосударственном масштабе использование малоэффективных или излишне дорогих лекарственных средств ведет к неоправданным расходам органов здравоохранения и отдельных потребителей, снижает результаты терапии или профилактических мер, подрывает доверие общества к производителям, к работникам аптечной сети и в целом к системе здравоохранения. Таким образом, пациент, как потребитель лекарственных средств, вынужден доверять всем: разработчику, исследователю, производителю, врачу, квалифицированная помощь которого может вовремя скорректировать назначение и т. д. Такая особенность лекарственного средства как специфического товара заставила установить требования к основным этапам их обращения, т. е. к разработке, испытаниям, регистрации, производству и т. д. Общепринятым является положение, что необходимые свойства лекарственного средства достигаются, когда лекарственное средство объединяет совокупность трех основных характеристик - эффективности, безопасности и качества. В последнем случае под качеством понимается более узкая категория (соответствие спецификации, монографии Фармакопеи, фармакопейной статье или другой аналитической нормативной документации), чем указанное выше КАЧЕСТВО лекарственного средства в глобальном смысле. Далее будут рассмотрены вопросы обеспечения КАЧЕСТВА лекарственного средства именно в глобальном смысле (рис. 1),кроме этого, мы будут рассмотрены механизмы, за счет которых такое КАЧЕСТВО достигается. 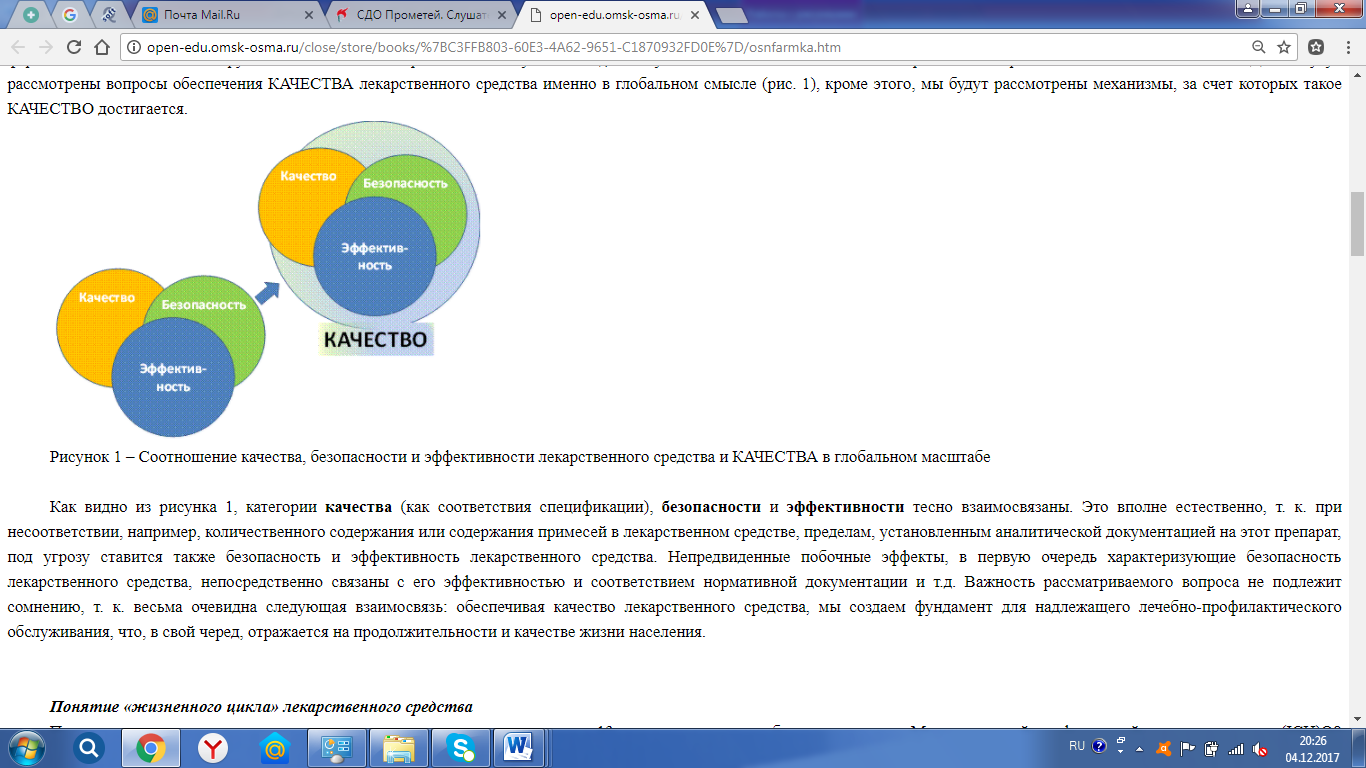 Рисунок 1 – Соотношение качества, безопасности и эффективности лекарственного средства и КАЧЕСТВА в глобальном масштабе Как видно из рисунка 1, категории качества (как соответствия спецификации), безопасности и эффективности тесно взаимосвязаны. Это вполне естественно, т. к. при несоответствии, например, количественного содержания или содержания примесей в лекарственном средстве, пределам, установленным аналитической документацией на этот препарат, под угрозу ставится также безопасность и эффективность лекарственного средства. Непредвиденные побочные эффекты, в первую очередь характеризующие безопасность лекарственного средства, непосредственно связаны с его эффективностью и соответствием нормативной документации и т.д. Важность рассматриваемого вопроса не подлежит сомнению, т. к. весьма очевидна следующая взаимосвязь: обеспечивая качество лекарственного средства, мы создаем фундамент для надлежащего лечебно-профилактического обслуживания, что, в свой черед, отражается на продолжительности и качестве жизни населения. Понятие «жизненного цикла» лекарственного средства Понятие «жизненного цикла» лекарственного средства появилось около 13 лет назад при разработке документов Международной конференцией по гармонизации (IСН)Q8 «Фармацевтическая разработка» и Q9 «Управление риском для качества». В контексте этих документов, принятых IСН в 2004-2005 гг., под «жизненным циклом» понимаются все стадии существования лекарственного средства от его разработки, нахождения в обращении на рынке и до прекращения его пребывания на рынке. В более позднем документе IСН Q10 «Фармацевтическая система качества» под «жизненным циклом» лекарственного средства уже понимаются: его разработка, внедрение (перенос технологий производства лекарственного средства на промышленный масштаб), промышленное производство и прекращение производства препарата. Отметим, что документ IСН Q10 является при этом рекомендательным, а не обязательным. Особо следует отметить то, что в документе IСН Q10 не рассматриваются вопросы обеспечения КАЧЕСТВА лекарственного средства на всех характерных этапах его обращения. Важно отметить, что «жизненный цикл» лекарственного средства не следует замыкать только на «рыночной» составляющей (рис.2)  Рисунок 2 – «Рыночные» составляющие жизненного цикла лекарственного средства Правильнее рассматривать «жизненный цикл» лекарственного средства на этапах его обращения. Они, в свою очередь, связаны с его медицинским или ветеринарным применением с целью диагностики, профилактики либо лечения заболеваний (возможно - изменения состояния и/или функций организма). Исходя их этого, «жизненный цикл» лекарственного средства охватывает все этапы, начиная от разработки, его доклинических исследований, клинических испытаний для доказательства эффективности и безопасности, включает промышленное производство лекарственного средства после его государственной регистрации, дистрибьюцию (или дистрибуцию – написание транскрипции в этих двух вариантах равнозначно) (в т. ч. транспортирование и хранение) и розничную реализацию с дальнейшим медицинским применением. Рассмотренная цепочка как раз и характеризует «жизненный цикл» лекарственного средства, начиная от его разработки (т. е. «рождения» лекарственного средства), и заканчивая его использованием пациентом (т. е. медицинским применением). 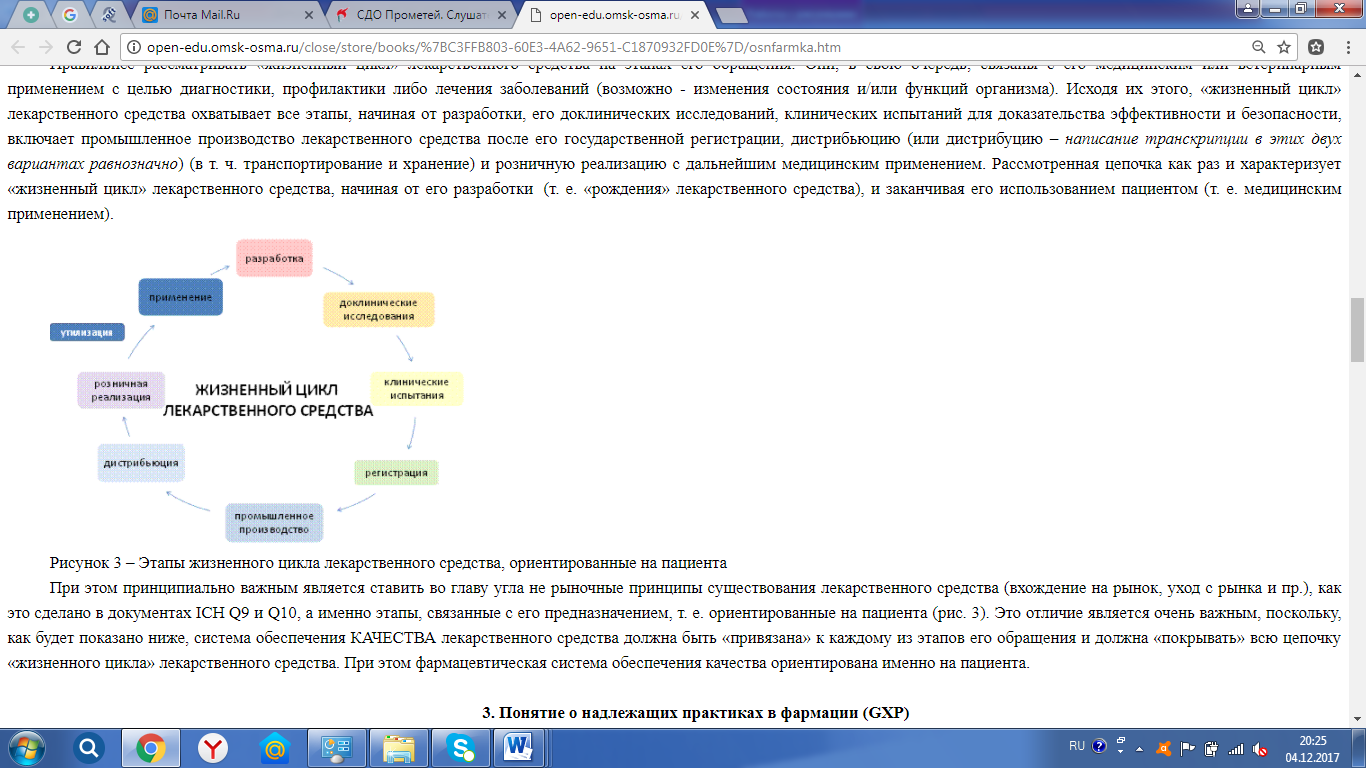 Рисунок 3 – Этапы жизненного цикла лекарственного средства, ориентированные на пациента При этом принципиально важным является ставить во главу угла не рыночные принципы существования лекарственного средства (вхождение на рынок, уход с рынка и пр.), как это сделано в документах IСН Q9 и Q10, а именно этапы, связанные с его предназначением, т. е. ориентированные на пациента (рис. 3). Это отличие является очень важным, поскольку, как будет показано ниже, система обеспечения КАЧЕСТВА лекарственного средства должна быть «привязана» к каждому из этапов его обращения и должна «покрывать» всю цепочку «жизненного цикла» лекарственного средства. При этом фармацевтическая система обеспечения качества ориентирована именно на пациента. 3. Понятие о надлежащих практиках в фармации (GХР) Примерно 15 лет назад группой ученых была сформулирована концепция глобального обеспечения качества на всех этапах обращения лекарственного средства, представленная на рисунке 4.На ней показаны этапы «жизненного цикла» лекарственного средства и соответствующие стандарты, которые устанавливают требования к системе обеспечения качества, применяемой на каждом этапе «жизненного цикла» лекарственного средства. Кратко охарактеризуем каждый из этапов. Первое, что мы должны помнить, это то, что лекарственное средство (ЛС) предназначено для пациента, поэтому его разработка инициируется, исходя из нужд системы здравоохранения. Это может быть бюджетный или социальный заказ или маркетинговая проработка компанией. Однако суть инициирования заключается в том, что компания будет разрабатывать (а в дальнейшем и выпускать) только то лекарственное средство, которое будет востребовано, и производство которого будет востребовано и принесет компании прибыль. Здесь мы видим ориентацию на пациента как на ключевое звено всего дальнейшего «жизненного цикла» лекарственного средства.  Рисунок 4 – Система обспечения качества на всех этапах обращения (жизненного цикла) лекарственного средства В начале III тысячелетия появилась концепция обеспечения качества лекарственного средства, согласно которой КАЧЕСТВО лекарственного средства не может быть достигнуто на этапе производства и подтверждено выборочным контролем. Это КАЧЕСТВО должно «встраиваться» в лекарственное средство, начиная с этапа его разработки, а затем уже воспроизводиться при промышленном производстве и поддерживаться при дистрибьюции. Результаты контроля качества лекарственного средства при этом воспринимаются только как дополнительное подтверждение соответствия продукции установленным при государственной регистрации требованиям. К процессу разработки лекарственного средства, так же как и к другим этапам обращения, или «жизненного цикла» лекарственного средства, представленного на рисунке 4, применяются специфические для этого этапа нормативные требования. Они изложены в документе IСН Q8 «Фармацевтическая разработка», который был принят в 2005 г. Документ IСНQ8 - не только руководство для производителей лекарственных средств, но он также может эффективно использоваться при проведении экспертизы регистрационных материалов на лекарственное средство в процессе государственной регистрации. Этапы доклинических и клинических исследований лекарственных средств, необходимые для подтверждения их эффективности и безопасности, проводятся в соответствии с требованиями надлежащих практик: лабораторной (GLР) и клинической (GСР). Требования надлежащей лабораторной практики - это специфическая система качества, которая является обязательной при проведении доклинических исследований лекарственного средства. Они были разработаны в США, рекомендованы ВОЗ, затем приняты на уровне организации экономического сотрудничества и развития ОЕСD и имплементированы в законодательства стран ЕС. Считается, что проведение исследований в соответствии с этой системой качества обеспечивает платформу для взаимного признания их результатов. Требования GСР распространяются на проведение клинических исследований лекарственных средств и являются обязательными. Они были приняты на уровне IСН и также имплементированы в законодательство стран ЕС как обязательные нормативные документы. В процессе государственной регистрации лекарственного средства как раз и происходит экспертная оценка лекарственного средства на предмет его эффективности, безопасности и качества, при этом рассматриваются документальные подтверждения этих характеристик лекарственного средства. После того, как лекарственное средство зарегистрировано (при этом не имеет значения, регистрируется ли оно для внутреннего рынка или для экспорта), начинается его промышленный выпуск. На этом этапе действует отдельная специфическая система обеспечения качества лекарственного средства - правила надлежащего производства (GМР). Принципы GМР обычно утверждаются на уровне Законов, а детальные руководства (правила) - на уровне подзаконных актов. Соблюдение и тех, и других является обязательным условием получения лицензии на производство лекарственного средства и осуществление такого производства. Основная направленность GМР - это обеспечение однородности серий, стабильности и воспроизводимости производственных процессов и процедур, их соответствия регистрационной документации, в т. ч. соответствие показателей лекарственного средства утвержденной спецификации. Таким образом, правила GМР затрагивают исключительно фармацевтические аспекты качества препаратов. В настоящее время нельзя мгновенно улучшить КАЧЕСТВО ЛС при его производстве в соответствии с требованиями GМР. Это качество может быть именно таким, как предусмотрено в регистрационном досье, при этом гарантирована стабильность этого КАЧЕСТВА. Если же мы хотим получить «лучший» препарат, например, с модифицированным высвобождением для обеспечения пролонгированного действия, то это будет уже не зарегистрированный ранее, а другой препарат, который будет проходить все ранее описанные этапы разработки, исследования и регистрации. При дальнейшем хранении и дистрибьюции КАЧЕСТВО лекарственного средства должно поддерживаться при соблюдении правил надлежащего хранения (GSР) и дистрибьюции(GDР) лекарственного средства. При этом правила надлежащей дистрибьюции обычно вводятся в лицензионные требования к осуществлению оптовой реализации лекарственного средства. На этапе розничной реализации, когда оно уже доходит до пациента, существуют правила надлежащей фармацевтической или аптечной практики, помогающие отпустить пациенту именно те КАЧЕСТВЕННЫЕ лекарственные средства, в которых он нуждается. Существуют и другие многочисленные правила, обобщающий термин для которых получил название «GХР», однако при всем многообразии «надлежащих практик» необходимо иметь в виду то, что каждая из них является системой обеспечения КАЧЕСТВА лекарственного средства на определенном этапе его «жизненного цикла». Для обозначения различных «практик» уже начинает ощущаться нехватка сокращений. Так, аббревиатура GРР употребляется для обозначения Правил надлежащей закупки (Gооd Procurement Ргасtiсе), Правил надлежащей фармацевтической практики (Gооd Pharmacy Ргасtiсе), Правил публикационной практики (Gооd Publication Ргасtiсе), что может вызвать путаницу. В целях сокращения совокупность различных «практик» обозначают аббревиатурой GХР, где «X» может заменять C, D, L М, Р и др. (таблица 1). Таблица 1 – Общепринятые русскоязычные эквиваленты названия надлежащих практик
Таким образом, можно говорить о цепочке обеспечения качества, охватывающей весь «жизненный цикл» лекарственного средства, суть которой заключается в ориентации на пациента, последовательности и непрерывности (рис. 4, 5).Надлежащие лабораторная, клиническая, производственная практика (GLР, GСР, GМР) и надлежащая практика дистрибьюции (GDР) принимаются обычно на уровне законодательства стран со строгой регуляторной системой. Если оперировать терминами системы стандартизации, можно было бы назвать эти документы основополагающими (другими словами - системообразующими) стандартами. Правила взаимозависимы и в сфере применения. Так, Правила GМР требуют, чтобы новые препараты, передаваемые в серийное производство, были разработаны и испытаны в соответствии с Правилами GLР и GСР. В свою очередь, предусматривается, что биохимические и другие лаборатории, участвующие в доклинических испытаниях препаратов, должны отвечать требованиям СLР. Аналогичные требования все шире распространяются и на аналитические лаборатории, занятые контролем качества лекарств.  Рисунок 5 – Взаимосвязь правил надлежащей практики Отражая различные аспекты единой концепции обеспечения качества, эффективности и без опасности лекарств, Правила GМР, GСР и СLР тесно связаны между собой внутренней логикой и подходами. Эти три свода правил основаны на комплексном учете и недопущении всех факторов, способных отрицательно повлиять на качество лекарственного средства. Кроме этого, трансфер (масштабирование) технологий в промышленных условиях, а также наработка препаратов на клинические испытания должны быть выполнены в соответствии с требованиями GМР. Лишь в этом случае можно гарантировать неизменность характеристик и свойств лекарственного средства, подтверждающие их эффективность и безопасность, с одной стороны (лекарственные средства на этапе исследований), и, с другой стороны - лекарственные средства, которые выпускаются в промышленных условиях. Вместе с тем между этими сводами правил имеются различия, прежде всего в отношении сферы применения, которые суммированы в таблице 2. Таблица 2 - Различия между GLP, GCP, GMP
В настоящее время на территории Российской Федерации законодательно утверждены и применяются: Правила надлежащей производственной практики - утверждены приказом Минпромторга России 14 июня 2013 г. N 916 (в ред. Приказа Минпромторга России от 18.12.2015 N 4148). Правила организации и проведения инспектирования производителей лекарственных средств на соответствие требованиям правил надлежащей производственной практики, а также выдачи заключений о соответствии производителя лекарственных средств указанным требованиям» - утверждены Постановлением Правительства РФ от 3 декабря 2015 года №1314. Приказ Министерства здравоохранения Российской Федерации № 199н от 01.04.2016 г. «Об утверждении Правил надлежащей лабораторной практики». Приказ Министерства здравоохранения Российской Федерации от 31.08.2016 № 647н «Об утверждении Правил надлежащей аптечной практики лекарственных препаратов для медицинского применения». Вопросы по материалам лекции: 1. Дать определение термина «качество». 2. Дать определение термина «качество лекарственного средства». 3. Перечислить особенности рецептурного лекарственного препарата как товара. 4. Перечислить (в строгой последовательности) этапы «жизненного цикла» лекарственного средства. 5. Дать определение концепции глобального обеспечения качества лекарственных средств. 6. Привести русскоязычный эквивалент GMP. 7. Привести русскоязычный эквивалент GLP. 8. Привести русскоязычный эквивалент GCP. 9. Привести русскоязычный эквивалент GDP. 10. Привести русскоязычный эквивалент GPP. 11. Привести русскоязычный эквивалент GSP. 12. Привести русскоязычный эквивалент GPvP. 13. Перечислить отличия правил GMP по критериям: - вид деятельности; - значение для препаратов; - обеспечение свойств препаратов - этические аспекты. |