|
|
ЛЕКЦИЯ 14. Лекция 14 Электрокинетические свойства коллоидных систем. Электрофорез, электроосмос. Строение коллидных частиц лиофобных золей, электрокинетический потенциал.
ЛЕКЦИЯ 14
Электрокинетические свойства коллоидных систем. Электрофорез, электроосмос. Строение коллидных частиц лиофобных золей, электрокинетический потенциал. Влияние электролитов на величину электрокинетического потенциала. Изоэлектрическое состояние. Устойчивость и коагуляция коллоидных систем. Коагуляция электролитами, правило Шульце-Гарди.
14.1. Строение коллоидных частиц лиофобных золей, электрокинетический потенциал.
Согласно общепринятой мицеллярной теории строения коллоидных растворов, золь (коллоидный раствор) состоит из двух частей: мицелл и интермицеллярной жидкости.
Мицелла – это частица дисперсной фазы золя, окруженная двойным электрическим слоем.
Интермицеллярной (межмицеллярной) жидкостью называют дисперсионную среду, разделяющую мицеллы, в которой растворены электролиты, неэлектролиты и ПАВ, являющиеся стабилизаторами коллоидной системы.
Мицеллы
Частицы дисперсной фазы золей называют мицеллами. Если исключить влияние растворителя, в котором образуется коллоидная система, то упрощенную схему строения мицеллы золя хлорида серебра (при избытке хлорид-анионов) можно представить следующим образом. Предположим, что золь хлорида серебра получен сливанием сильно разбавленных растворов хлорида калия и нитрата серебра, причем хлорид калия взят в избытке.
При взаимодействии катионов серебра с хлорид-анионами образуются частицы нерастворимого в воде хлорида серебра. Поскольку растворы сильно разбавлены, микрокристаллы получаются коллоидных размеров, очень мелкие. Такой микрокристалл образует ядро мицеллы.
Рост кристалла прекращается, когда в растворе практически до нуля падает концентрация ионов серебра. Но хлорид-анионы присутствуют в избытке. Часть из них адсорбируется на поверхности ядра, достраивая его кристаллическую решетку. Хлорид-анионы в данном случае называют потенциалопределяющими ионами. Именно они обусловливают наличие отрицательного заряда агрегата ядра с избытком ионов С1-. Если бы в растворе присутствовал избыток нитрата серебра, потенциалопределяющими ионами были бы катионы Ag+.
Естественно, после возникновения заряда образовавшаяся частица начинает притягивать из раствора ионы с противоположным знаком - катионы калия (противоионы), образуется так называемый двойной электрический слой. Некоторая часть противоионов очень прочно притягивается к агрегату, образуя адсорбционный слой. Часть мицеллы, включающую ядро, потенциал определяющие ионы и адсорбционный слой, называют гранулой. Ионы К+, которые не входят в адсорбционный слой, слабее связаны с гранулой и могут диссоциировать в раствор. Они составляют диффузный слой противоионов.
В целом мицелла представляет собой электронейтральную частицу, но за счет перехода части ионов диффузного слоя в раствор гранулы имеют на поверхности избыточный отрицательный заряд, который и препятствует их коагуляции в более крупные частицы.
Строение мицеллы можно изобразить с помощью формулы. Последовательные шаги в составлении формулы мицеллы таковы.
Ядро мицеллы состоит из т частиц AgCl, образующих микрокристалл: m[AgCl].
Потенциалопределяющие ионы адсорбируются на поверхности ядра; предположим, что для нашего примера их число равно п: m[AgCl] • nСl-.
Затем следует слой противоионов. Их общее число так же равно п, однако часть (допустим, х) из них образуют диффузный слой, остальные (п - х) вместе с ядром и потенциалопределяющими ионами составляют гранулу. Часть формулы, относящуюся к грануле мицеллы, заключают в фигурные скобки. Заряд гранулы в данной мицелле равен х . Таким образом, формула мицеллы золя хлорида серебра в избытке хлорид-анионов такова:
{m[AgCl] nCl- • (п - х)К+}-х хК+
При этом основу коллоидных частиц составят микрокристаллы труднорастворимого AgCI, включающие в себя m молекул AgCI (а точнее, m пар ионов Ag+ и CI). Эти микрокристаллы называют агрегатами. Если реакция протекает в присутствии избытка иодида калия, то на поверхности агрегата возникает отрицательно заряженный слой в результате избирательной адсорбции n ионов CI. Иодид-ионы являются потенциалобразующими ионами (сокращенно ПОИ). Агрегат вместе с потенциалобразующими ионами является частицей твердой фазы и его называют ядром.
Под действием электростатических сил к ядру притягивается n ионов противоположного знака – противоионов, компенсирующих заряд ядра. В данном случае эту роль выполняют ионы K+. Часть противоионов (n - x), наиболее близко расположенных к ядру, находится в слое жидкости, смачивающем поверхность твердого ядра. Эти ионы испытывают действие не только электростатических, но и ван-дер-ваальсовых сил ядра, поэтому прочно удерживаются около него и образуют так называемый адсорбционный слой противоионов. Ядро с адсорбционным слоем противоионов образует коллоидную частицу. Остальные x противоионов, слабее связанных с ядром (только электростатически), под влиянием теплового движения располагаются в жидкой фазе диффузно (размыто), почему и носят название диффузного слоя. Все это образование вместе и является мицеллой.
Мицеллы золей электронейтральны.
Числа m, n и x могут изменяться в широких пределах в зависимости от условий получения и очистки золя. Обычно m >> n. Ядро вместе с адсорбционным слоем противоионов образуют собственно коллоидную частицу, или гранулу. В отличие от электронейтральной мицеллы коллоидная частица имеет заряд, в данном случае отрицательный (x ).
Граница между коллоидной частицей и диффузным слоем носит название поверхность скольжения. В формуле мицеллы этой границе соответствует фигурная скобка между адсорбционным и диффузным слоями (на рис. 1 сплошная линия).
Пример строения мицеллы для иодида серебра.

Рис. 1. Схема строения мицеллы золя иодида серебра
с отрицательно заряженными частицами.
Граница скольжения обозначает ту геометрическую поверхность, по которой происходит разделение («разрыв») мицеллы на коллоидную частицу и диффузный слой в случае ее перемещения относительно дисперсионной среды (например, при участии мицеллы в броуновском движении или при движении под действием электрического поля).
На границе раздела твердое тело – жидкость возникает двойной электрический слой. Согласно современным представлениям, двойной электрический слой (ДЭС) это образующийся на границе двух фаз тонкий поверхностный слой из пространственно разделенных электрических зарядов противоположного знака (потенциалобразующих ионов и противоионов). Потенциалобразующие ионы, адсорбирующиеся на твердой поверхности, это внутренняя обкладка ДЭС. Внешняя обкладка ДЭС (слой противоионов) состоит из двух частей: плотной и диффузной.
Образование двойного слоя ионов приводит к появлению определенных электрических потенциалов на границе раздела твердой и жидкой фаз. Ионы первого слоя (внутренней обкладки), фиксированные на твердой поверхности, придают этой поверхности свой знак заряда и создают на ней так называемый поверхностный или φ-потенциал. Знак φ-потенциала совпадает со знаком заряда потенциалобразующих ионов. Величина φ-потенциала пропорциональна числу зарядов этих ионов на поверхности частиц.
С точки зрения термодинамики, φ-потенциал равен работе переноса единичного (элементарного) заряда из бесконечно удаленной точки объема раствора на поверхность твердой фазы, т. е. представляет собой потенциал твердой поверхности. Прямых методов его измерения не имеется.
Второй потенциал, характеризующий двойной слой ионов, называют электрокинетическим потенциалом или -потенциалом (дзета-потенциалом). Он представляет собой электрический потенциал в двойном слое на границе между коллоидной частицей, способной к движению в электрическом поле и окружающей жидкостью. -потенциал является потенциалом поверхности скольжения. Однако в двойном электрическом слое точное расстояние от твердой поверхности до поверхности скольжения неизвестно. Поэтому приближенно можно принять, что поверхность скольжения проходит по границе между адсорбционным и диффузным слоями противоионов. Следовательно -потенциал близок, хотя и не совсем равен, потенциалу на границе адсорбционного и диффузионного слоев.
Термодинамически ξ-потенциал можно определить как работу, необходимую для переноса единичного заряда из бесконечно удаленного элемента объема раствора на поверхность скольжения. Знак ξ-потенциала обычно совпадает со знаком φ-потенциала. ξ-потенциал является частью φ потенциала и всегда меньше, чем φ потенциал. Величина ξ-потенциала непосредственно связана с числом противоионов в диффузном слое и изменяется пропорционально этому числу. Можно считать, что с увеличением толщины диффузного слоя ξ потенциал повышается. Поскольку электрокинетический потенциал относится к коллоидной частице и обусловливает ее подвижность в электрическом поле, величина этого потенциала может быть измерена экспериментально по скорости движения частиц. Направление же перемещения частиц к катоду или аноду указывает на знак ξ-потенциала.
Благодаря наличию ξ-потенциала на границах скольжения всех частиц дисперсной фазы возникают одноименные заряды и электростатические силы отталкивания противостоят процессам агрегации. Таким образом, ξ-потенциал является одним из основных факторов агрегативной устойчивости гидрофобных золей. Величина, а иногда и знаки φ- и ξ-потенциалов могут изменяться под влиянием внешних воздействий (электролитов, разведения, повышения температуры). Особенно чувствителен к этим факторам ξ-потенциал.
Виды устойчивости дисперсных систем. Лиофобные и лиофильные золи
Устойчивость дисперсных систем – это возможность их нахождения в исходном состоянии неопределенно долгое время.
Устойчивость дисперсных систем может быть:
К осаждению дисперсной фазы - характеризует способность дисперсной системы сохранять равновесное распределение фазы по объему дисперсионной среды или ее устойчивость к разделению фаз. Это свойство называется седиментационная (кинетическая) устойчивость.
К агрегации ее частиц.
Агрегативная устойчивость – это способность дисперсной системы сохранять неизменной во времени степень дисперсности, т.е. размеры частиц и их индивидуальность.
Она обусловлена способностью дисперсных систем образовывать агрегаты (т.е. укрупняться). По отношению к агрегации дисперсные системы могут быть устойчивыми кинетически и термодинамически. Термодинамически устойчивые системы образуются в результате самопроизвольного диспергирования одной из фаз, т.е. самопроизвольного образования гетерогенной свободнодисперсной системы. Дисперсные системы также делят на:
Особенности этих двух видов устойчивости показаны на схеме:
Устойчивость
дисперсных систем
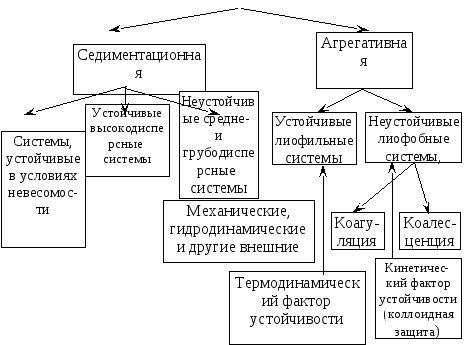
Термодинамическая устойчивость лиофильных систем означает, что они равновесны (энергия Гиббса G min), обратимы и образуются самопроизвольно, как из макрофаз, так и из истинных растворов. Поскольку образуются гетерогенные системы, то поверхностная энергия должна быть скомпенсирована энтропийной составляющей, т.е. частицы дисперсной системы должны участвовать в молекулярно кинетическом (тепловом) движении. Отсюда следует, что лиофильные системы могут быть только ультромикрогетерогенными, а поверхностное натяжение на границе «частица – среда» должно быть очень малым. Значение поверхностного натяжения, при котором обеспечивается термодинамическая устойчивость дисперсных систем, определяется соотношением Ребиндера – Щукина:
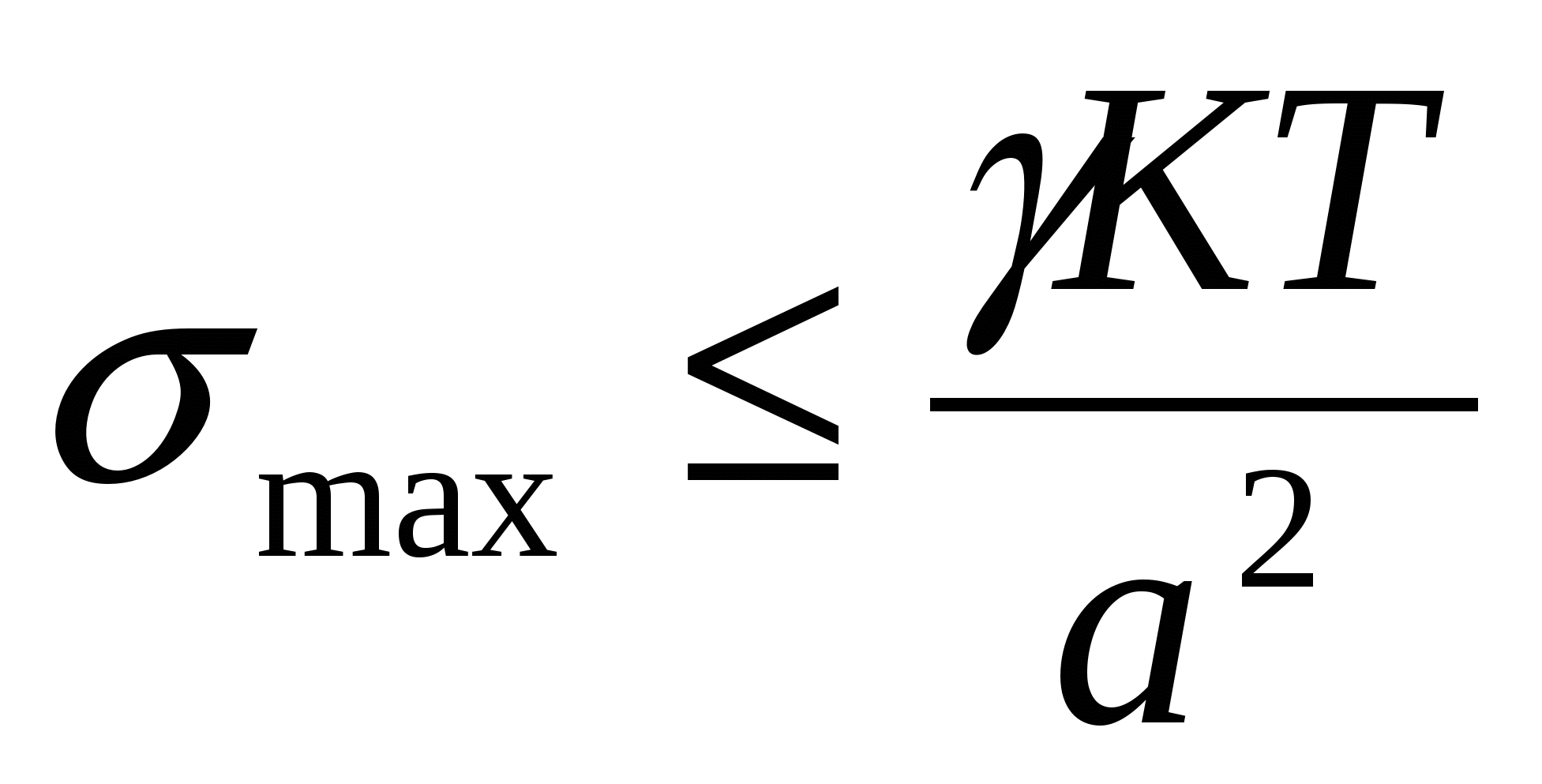 , ,
где ỵ езразмерный коэффициент;
K – постоянная Больцмана;
а – средний размер частицы.
Расчеты показывают, что межфазное поверхностное натяжение в лиофильных дисперсных системах в зависимости от размеров частиц может иметь значение от 1,410-7 до 1,410-3 Дж/м2. Типичными представителями лиофильных дисперсных систем являются растворы коллоидных поверхностно активных веществ (ПАВ) (ассоциативные коллоиды) и растворы полимеров (молекулярные коллоиды).
Лиофобные системы термодинамически неустойчивы, т.к. частицы дисперсной фазы склонны к агрегации. Их агрегативная термодинамическая неустойчивость обусловлена избытком поверхностной энергии. Межфазное натяжение в них больше рассчитанного по соотношению Ребиндера – Щукина, поэтому они не могут быть получены самопроизвольным диспергированием. Для их образования должна быть затрачена внешняя энергия. Укрупнение частиц дисперсной фазы при потере агрегативной устойчивости достигается двумя путями:
Изотермическая перегонка, т.е. растворение мелких и рост крупных частиц в соответствии с уравнением Кельвина;
За счет слипания частиц, т.е. коагуляцией.
В зависимости от природы среды и концентрации дисперсной фазы эти процессы могут заканчиваться или осаждением, или структурообразованием.
При нарушении агрегативной устойчивости происходит коагуляция.
Правила коагуляции электролитами. Порог коагуляции. Правило Шульце-Гарди. Виды коагуляции: концентрационная и нейтрализационная. Коагуляция смесями электролитов. Явление «неправильные ряды». Механизм и кинетика коагуляции
Коагуляцией называется процесс слипания частиц с образованием крупных агрегатов. В результате коагуляции система теряет свою седиментационную устойчивость, так как частицы становятся слишком крупными и не могут участвовать в броуновском движении.
Коагуляция является самопроизвольным процессом, так как она приводит к уменьшению межфазной поверхности и, следовательно, к уменьшению свободной поверхностной энергии.
Различают две стадии коагуляции.
1 стадия – скрытая коагуляция. На этой стадии частицы укрупняются, но еще не теряют своей седиментационной устойчивости.
2 стадия - явная коагуляция. На этой стадии частицы теряют свою седиментационную устойчивость. Если плотность частиц больше плотности дисперсионной среды, образуется осадок.
Причины коагуляции многообразны. Едва ли существует какое либо внешнее воздействие, которое при достаточной интенсивности не вызывало бы коагуляцию.
|
|
|
 Скачать 0.76 Mb.
Скачать 0.76 Mb.