ЛЕКЦИЯ 14. Лекция 14 Электрокинетические свойства коллоидных систем. Электрофорез, электроосмос. Строение коллидных частиц лиофобных золей, электрокинетический потенциал.
 Скачать 0.76 Mb. Скачать 0.76 Mb.
|

При коагуляции смесью электролитов различают два типа процессов:
Гомокоагуляция - укрупнение подобных частиц в больший агрегат осадка. Причем в процессе отстаивания мелкие частицы растворяются, а крупные увеличиваются за их счет. На этом основано явление активации и перекристаллизации. Этот процесс описывается уравнением Кельвина – Томсона: где С - растворимость макрочастиц; С – растворимость микрочастиц; Vм – молярный объем; R – универсальная газовая постоянная; T – температура; r – радиус частиц. Из уравнения следует, что концентрация вокруг маленького радиуса больше, поэтому диффузия идет от бóльшей концентрации к меньшей. При втором типе происходит слияние разнородных частиц или прилипание частиц дисперсной системы на вводимые в систему чужеродные тела или поверхности. 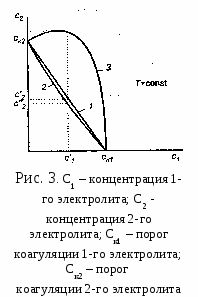 Гетерокоагуляция - взаимная коагуляция разнородных дисперсных систем. Коагуляция смесью электролитов имеет большое практическое значение, так как даже при добавлении к золю одного электролита-коагулянта, в действительности коагуляция происходит, по крайней мере, под влиянием двух электролитов, так как в системе содержится электролит-стабилизатор. Кроме того, в технике для коагуляции часто применяют смесь двух электролитов. Понимание закономерностей взаимного действия электролитов важно также при исследовании воздействия биологически активных ионов на органы и ткани живого организма. При коагуляции золя смесью двух и более электролитов возможны три случая (рис. 3.1.2.3). По оси абсцисс отложена концентрация первого электролита С1, а Cк1 – его порог коагуляции. Аналогично по оси ординат отложена концентрация второго электролита С2, а Ск2 – его порог коагуляции. 1. Аддитивное действие электролитов (линия 1 рис.3). Электролиты действуют как бы независимо один от другого, их суммарное действие складывается из воздействий каждого из электролитов. Если с1´ - концентрация первого электролита, то для коагуляции золя концентрация второго электролита должна быть равной с2´. Аддитивность наблюдается обычно при сходстве коагулирующей способности обоих электролитов. 2. Синергизм действия (линия 2 рис..3). Электролиты как бы способствуют друг другу – для коагуляции их требуется меньше, чем нужно по правилу аддитивности (с2″ < c2′). Условия, при которых наблюдается синергизм, сформулировать трудно. 3. Антагонизм действия (линия 3 рис.3). Электролиты как бы противодействуют друг другу и для коагуляции их следует добавить больше, чем требуется по правилу аддитивности. Антагонизм наблюдается при большом различии в коагулирующем действии электролитов. Существует несколько теорий, объясняющих явление антагонизма. Одной из его причин может служить химическое взаимодействие между ионами. Например, для золя AgCl, стабилизированного хлоридом калия, коагулирующем действием обладают катионы. Например, большой коагулирующей способностью обладает четырёхзарядный ион тория Th4+. Однако если взять для коагуляции смесь Th(NO3)4 и K2SO4, то коагулирующая способность этой смеси значительно меньше, чем отдельно взятого Th(NO3)4. Связано это с тем, что в результате химической реакции образуется комплекс: и вместо четырёхзарядных ионов Th4+ в золе будут находиться однозарядные катионы K+, коагулирующее действие которых значительно слабее (правило Шульце-Гарди). Гетероадагуляция - прилипание частиц дисперсной фазы к вводимой в систему чужеродной поверхности. Одной из причин этого явления является адсорбция стабилизатора на этой поверхности. Например: отложение коллоидных частиц на волокнах при крашении и дроблении. Для гидрофобных золей в качестве ВМС обычно используют белки, углеводы, пектины; для неводных золей – каучуки. При введении в коллоидный раствор электролитов, содержащих многовалентные ионы с зарядом противоположные заряду частиц, наблюдается явление «неправильные ряды». Оно состоит в том, что при добавлении к отдельным порциям золя все возрастающего его количества электролита золь сначала остается устойчивым, затем в определенном интервале концентраций происходит коагуляция; далее золь снова становится устойчивым и, наконец, при повышении концентрации электролита опять наступает коагуляция уже окончательная. Подобные явления могут вызывать и большие органические ионы. Объясняется это тем, что при весьма малых количествах введенного электролита ионов недостаточно, чтобы коагулировать золь, т. е. значение - потенциала остается выше привычного (рис. 4). При больших количествах электролита его ионы проявляют коагулирующее действие. Этот интервал концентраций отвечает значениям - потенциала частиц от критического первого знака до критического другого знака. 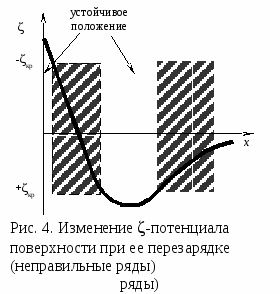 При еще больших концентрациях многовалентные ионы перезаряжают коллоидную частицу и золь опять устойчивый. В этой зоне -потенциал опять выше критического значения, но имеет обратный по знаку частицам заряд исходного золя. Наконец, при высоком содержании исходного электролита многовалентные ионы снова снижают значение -потенциала ниже критического и снова происходит окончательная коагуляция. Повышение агрегативной устойчивости золя путём введения в него высокомолекулярного соединения (ВМС) называется коллоидной защитой. Происходит образование защитной пленки на поверхности золя (гидратной или ВМС), препятствующей взаимодействию частиц электролита. В качестве количественной характеристики коагуляции Зигмонди предложил использовать скорость коагуляции. Скорость коагуляции - это изменение концентрации коллоидных частиц в единицу времени при постоянном объеме системы. где - концентрация частиц; - время. Знак «-» стоит потому, что концентрация частиц со временем уменьшается, а скорость всегда положительна. Степень коагуляции : где Z - общее число столкновений частиц в единицу времени; Zэф - число эффективных столкновений (т.е. столкновений, приводящих к коагуляции) в единицу времени. Если = 0, коагуляция не происходит, коллоидный раствор агрегативно устойчив. Если = 1, происходит быстрая коагуляция, т.е. каждое столкновение частиц приводит к их слипанию. Если 0 1, наблюдается медленная коагуляция, т.е. только некоторые столкновения частиц приводят к их слипанию. Чтобы частицы при столкновении слиплись, а не разлетелись как упругие шары, должен быть преодолен потенциальный барьер коагуляции ΔUк. Следовательно, коагуляция произойдет только в том случае, когда коллоидные частицы будут обладать кинетической энергией, достаточной дл преодоления этого барьера. Для увеличения степени коагуляции необходимо снижать потенциальный барьер. Это может быть достигнуто добавлением к золю электролита – коагулянта. Зависимость скорости коагуляции от концентрации электролита представлена на рис.5. На графике видны три участка: I. Следовательно, кинетическая энергия kТ << ΔUк, (k – постоянная Больцмана) – лиофобный золь агрегативно устойчив. II. 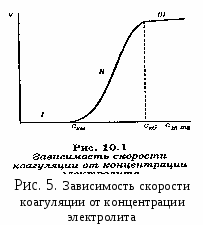 На этом участке происходит медленная коагуляция. III. Каждое столкновение приводит к слипанию частиц – идет быстрая коагуляция. Теория быстрой коагуляции, разработанная М. Смолуховским в 1916 г., основана на следующих положениях.
где k – константа скорости коагуляции. Проинтегрируем это уравнение, разделив переменные: где 0 – концентрация частиц золя в начальный момент времени; t – концентрация частиц золя в момент времени t. Для характеристики быстрой коагуляции используется период коагуляции(период половинной коагуляции) . Период коагуляции () – это время, через которое концентрация коллоидных частиц уменьшается в два раза. При Согласно теории быстрой коагуляции, константа коагуляции зависит от коэффициента диффузии и может быть вычислена по уравнению Если подставить в это уравнение величину коэффициента диффузии, получим: Таким образом, зная вязкость дисперсионной среды и температуру, можно вычислить константу скорости быстрой коагуляции. Теория Смолуховского неоднократно проверялась экспериментально и получила блестящее подтверждение, несмотря на сделанные автором допущения. Медленная коагуляция связана с неполной эффективностью столкновений вследствие существования энергетического барьера. Простое введение величины степени коагуляции в формулы теории Смолуховского не привело к согласию теории с опытом. Более совершенную теорию медленной коагуляции разработал Н.Фукс. Он ввел в кинетическое уравнение коагуляции множитель, учитывающий энергетический барьер коагуляции ΔU к: где kКМ – константа скорости медленной коагуляции; kКБ - константа скорости быстрой коагуляции; Р – стерический фактор; ΔUк - потенциальный барьер коагуляции; k – постоянная Больцмана. Таким образом, для расчета константы скорости медленной коагуляции необходимо знать потенциальный барьер коагуляции, величина которого зависит прежде всего от – потенциала. Фактор устойчивости, или коэффициент замедления W, показывает, во сколько раз константа скорости медленной коагуляции меньше константы скорости быстрой коагуляции. 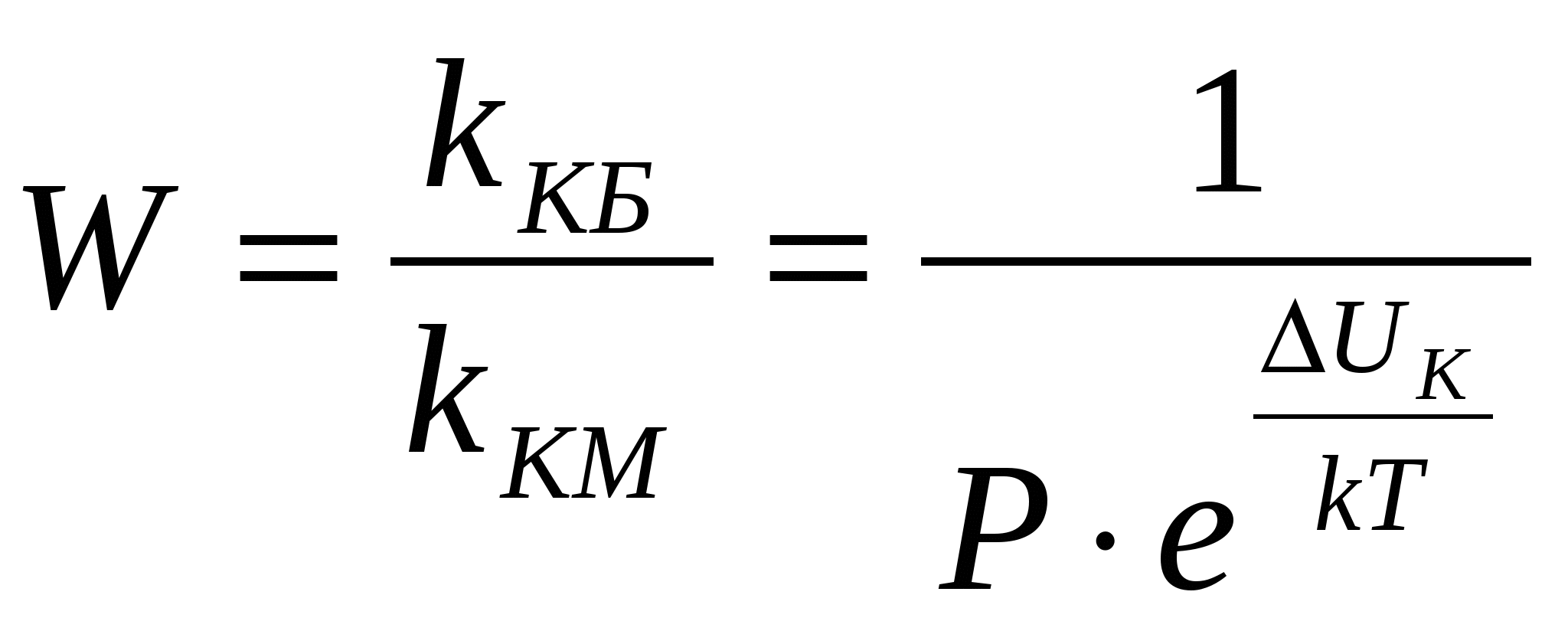 , ,Следует отметить пять факторов устойчивости, среди которых два первых играют главную роль.
Он обусловлен наличием ДЭС и – потенциала на поверхности частиц дисперсной фазы.
Он обусловлен снижением поверхностного натяжения в результате взаимодействия дисперсионной среды с частицей дисперсной фазы. Этот фактор играет заметную роль, когда в качестве стабилизаторов используются коллоидные ПАВ.
Он обусловлен тем, что на поверхности частиц дисперсной фазы образуются пленки, обладающие упругостью и механической прочностью, разрушение которых требует времени и затраты энергии. Этот фактор устойчивости реализуется в тех случаях, когда в качестве стабилизаторов используются высокомолекулярные соединения (ВМС).
Коагуляция приводит к уменьшению числа частиц в системе, следовательно, к уменьшению энтропии (ΔS<0), а это приводит к увеличению свободной энергии системы ΔG>0. Поэтому система самопроизвольно стремится оттолкнуть частицы друг от друга и равномерно (хаотично) распределить по объему системы. Этим обусловлен энтропийный фактор устойчивости. Однако число частиц в коллоидном растворе по сравнению с истинным раствором такой же массовой концентрации гораздо меньше, поэтому роль энтропийного фактора невелика. Но если частицы стабилизированы веществами, обладающими длинными гибкими цепями (ВМС) и потому имеющими много конформаций, то при сближении таких частиц их защитные слои вступают во взаимодействие. Это взаимодействие непременно приводит к уменьшению числа возможных конформаций, а значит – к уменьшению энтропии. Поэтому система стремится оттолкнуть частицы друг от друга.
Ему способствует увеличение плотности и динамической вязкости дисперсионной среды. В реальных системах действуют, как правило, несколько факторов устойчивости. Каждому фактору соответствует специфический способ его нейтрализации. Это затрудняет создание общей теории устойчивости. Пока существуют лишь частные теории. Седиментация и диффузия. Гипсометрический закон. Седиментационно-диффузионное равновесие. Скорость седиментации Грубодисперсные системы под действием гравитационных сил будут оседать (седиментировать). В результате в системе устанавливается определенное равновесие распределения частиц по высоте. Коллоидные системы по устойчивости занимают промежуточное положение между истинными растворами (max) и грубодисперсными растворами (min). На каждую частицу дисперсной фазы действует 3 силы:
Сила седиментации будет результирующей между первой и второй силой Если где B – коэффициент трения; U – скорость седиментации. С при равновесии Fсед = Fтр ила трения, согласно закону Стокса  для сферических частиц Отсюда уравнение скорости оседания и радиуса частиц  Результатами седиментационного анализа может служить интервал радиусов частиц в данной системе, просто радиус частиц или доля фракций определенного радиуса. Способность к седиментации принято выражать через константу седиментации S, которая определяется скоростью седиментации: Для сферических частиц эта константа равна Из уравнения следует, что Sзависит как от размеров частиц, так и от природы среды. За единицу измерения S принят сведберг (сб), равный 1013 с. Часто для характеристики процесса седиментации используют удельный поток седиментации Iсед. Удельный поток седиментации – это число частиц, оседающих в единицу времени через сечение единичной площади, нормальное к направлению седиментации. Размерность: [iсед] = част/см2 * с. Из определения iсед следует: iсед = Uсед * v, где v – частичная концентрация частиц в дисперсной системе. Подставив в это уравнение Uсед, получим: Таким образом, удельный поток прямо пропорционален V, v, (ρ – ρо) и обратно пропорционален S. Подставив эти выражения в уравнение, получим Значит, в случае сферических частиц удельный поток прямо пропорционален квадрату радиуса и обратно пропорционален вязкости среды. Рассматривая процесс седиментации, мы не учитываем броуновского движения, в котором участвуют частицы. Следствием броуновского движения, является диффузия, которая стремится выровнять концентрацию частиц по всему объёму, в то время как седиментация приводит к увеличению концентрации в нижних слоях. Таким образом, наблюдается два противоположных потока: поток седиментации iсед и поток диффузии iдиф. В результате конкуренции этих потоков возможны три варианта: 1. Чтобы выполнилось это неравенство, значения Т и 2. т.е. Это условие должно выполняться, когда Т и 3. т.е. В системе имеет место седиментационно-диффузионное равновесие. Проинтегрируем это уравнение, разделив переменные: Примем где vo – концентрация частиц на дне сосуда; vh – концентрация частиц на высоте h от дна. Отсюда В этом случае система является седиментационно-устойчивой, но распределение частиц в ней не равномерное, а равновесное. Это распределение наблюдается, когда 10-5 < r < 10-3 см. Если сравнить седиментацию с учетом диффузии и без нее, то видно различие факторов обусловливающих кинетическую устойчивость. Эти факторы позволяют различать кинетическую седиментационную устойчивость (КСУ) и термодинамическое равновесие, которого не может быть при КСУ. Мерой КСУ является величина, обратная константе седиментации. Эта устойчивость обеспечивается гидродинамическими факторами: вязкостью и плотностью среды, плотностью и размерами частиц. КСУ измеряется в обратных сведбергах: обр. св. = 1013 с– 1. 14.2.Электрокинетические свойства коллоидных систем: электрофорез, электроосмос. К электрокинетическим явлениям относят эффекты, связанные либо с относительным движением двух фаз под действием постоянного электрического поля, либо с возникновением разности потенциалов при относительном смещении двух фаз, на границе между которыми существует двойной электрический слой. Электрокинетические явления подразделяют на две группы: прямые и обратные. К прямым относят те электрокинетические явления, которые возникают под действием внешнего электрического поля (электрофорез и электроосмос). Обратными называют электрокинетические явления, в которых при механическом перемещении одной фазы относительно другой возникает электрический потенциал (потенциал протекания и потенциал седиментации). Электрофорез и электроосмос были открыты Ф. Рейссом (1808). Он обнаружил, что если во влажную глину погрузить две стеклянные трубки, заполнить их водой и поместить в них электроды, то при пропускании постоянного тока происходит движение частичек глины к одному из электродов. Это явление перемещения частиц дисперсной фазы в постоянном электрическом поле было названо электрофорезом. В другом опыте средняя часть U-образной трубки, содержащей воду, была заполнена толченым кварцем, в каждое колено трубки помещен электрод и пропущен постоянный ток. Через некоторое время в колене, где находился отрицательный электрод, наблюдалось поднятие уровня воды, в другом опускание. После выключения электрического тока уровни воды в коленах трубки уравнивались. Это явление перемещения дисперсионной среды относительно неподвижной дисперсной фазы в постоянном электрическом поле названо электроосмосом. Позже Квинке (1859) обнаружил явление, обратное электроосмосу, названное потенциалом протекания. Оно состоит в том, что при течении жидкости под давлением через пористую диафрагму возникает разность потенциалов. В качестве материала диафрагм были испытаны глина, песок, дерево, графит. Явление, обратное электрофорезу, и названное потенциалом седиментации, было открыто Дорном (1878). При оседании частиц суспензии кварца под действием силы тяжести возникала разность потенциалов между уровнями разной высоты в сосуде. Все электрокинетические явления основаны на наличии двойного электрического слоя на границе твердой и жидкой фаз. Из описанных явлений электрофорез имеет наиболее широкое практическое применение. При электрофорезе происходит направленное перемещение частиц дисперсной фазы в электрическом поле постоянного тока к электроду, знак которого противоположен знаку заряда частиц. Подвижность частиц в электрическом поле обусловлена тем, что при наложении внешней разности потенциалов происходит разрыв двойного электрического слоя по границе скольжения и частица получает заряд, соответствующий ее ξ-потенциалу. Противоионы диффузного слоя перемещаются при этом к противоположному электроду. Очевидно, что скорость движения частиц дисперсной фазы пропорциональна величине их ξ потенциала. Наблюдая электрофоретическое движение частиц, можно определить знак и величину ξ потенциала коллоидной частицы. Величина ξ потенциала связана со скоростью электрофореза заряженных частиц зависимостью, названной уравнением Гельмгольца – Смолуховского: где k — коэффициент, зависящий от формы частиц (для сферических частиц k = 6, для цилиндрических k = 4); η – вязкость среды; υ – линейная скорость перемещения частиц (или границы золя); ε – относительная диэлектрическая проницаемость среды; Е – напряженность поля (градиент потенциала). Линейная скорость υ изменяется пропорционально напряженности поля Е, поэтому не может служить характеристикой частиц. В связи с этим введено понятие электрофоретической подвижности υэф., равной скорости движения частицы при единичном градиенте потенциала (Е = 1): Экспериментально найденные значения подвижностей часто оказываются меньше расчетных. Несовпадение этих величин объясняете в основном тем, что теория Гельмгольца Смолуховского не учитывает два явления: релаксационный эффект и электрофоретическое торможение. Первый из этих эффектов вызывается нарушением симметрии диффузного слоя вокруг частиц. Второй эффект обусловлен добавочным трением электрической природы при движении частиц и противоионов в противоположные стороны. Методы электрофореза имеют большое теоретическое и практическое значение. Знание величины ξ-потенциала позволяет судить об устойчивости коллоидного раствора, поскольку изменение его устойчивости, как правило, происходит пропоционально изменению электрокинетического потенциала. Электроосмос, как и электрофорез, получил широкое применение. Механизм электроосмоса заключается в следующем. Нерастворимый материал мембраны при контакте с жидкостью (водой) диссоциирует с поверхности, отщепляя в жидкость те или другие ионы. Возникает двойной электрический слой, внутренняя обкладка которого входит в состав твердой фазы, а противоионы диффузно располагаются в жидкости. При включении постоянного электрического тока противоионы диффузного слоя перемещаются к электроду соответствующего знака. Так как ионы в воде всегда гидратированы, то при движении иона с ним увлекается определенный объем дисперсионнной среды за счет сил молекулярного трения (вязкости) между гидратной оболочкой иона и окружающей жидкостью. Очевидно, что чем больше толщина диффузного слоя и меньше площадь поперечного сечения капилляра или поры мембраны, тем сильнее проявляется электроосмотический перенос жидкости. Метод электроосмоса имеет большое практическое применение в процессах обезвоживания и сушки многих пористых материалов или весьма концентрированных коллоидных систем. Дли этой цели применяют, например, электрофильтр-прессы. Виды устойчивости дисперсных систем. Лиофобные и лиофильные золи Устойчивость дисперсных систем – это возможность их нахождения в исходном состоянии неопределенно долгое время. Устойчивость дисперсных систем может быть:
Агрегативная устойчивость – это способность дисперсной системы сохранять неизменной во времени степень дисперсности, т.е. размеры частиц и их индивидуальность. Она обусловлена способностью дисперсных систем образовывать агрегаты (т.е. укрупняться). По отношению к агрегации дисперсные системы могут быть устойчивыми кинетически и термодинамически. Термодинамически устойчивые системы образуются в результате самопроизвольного диспергирования одной из фаз, т.е. самопроизвольного образования гетерогенной свободнодисперсной системы. Дисперсные системы также делят на:
Особенности этих двух видов устойчивости показаны на схеме: 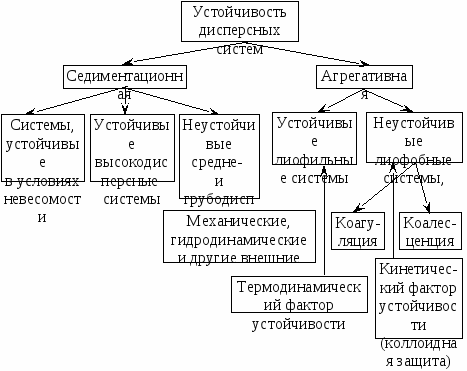 Термодинамическая устойчивость лиофильных систем означает, что они равновесны (энергия Гиббса G min), обратимы и образуются самопроизвольно, как из макрофаз, так и из истинных растворов. Поскольку образуются гетерогенные системы, то поверхностная энергия должна быть скомпенсирована энтропийной составляющей, т.е. частицы дисперсной системы должны участвовать в молекулярно кинетическом (тепловом) движении. Отсюда следует, что лиофильные системы могут быть только ультромикрогетерогенными, а поверхностное натяжение на границе «частица – среда» должно быть очень малым. Значение поверхностного натяжения, при котором обеспечивается термодинамическая устойчивость дисперсных систем, определяется соотношением Ребиндера – Щукина: где ỵ езразмерный коэффициент; K – постоянная Больцмана; а – средний размер частицы. Расчеты показывают, что межфазное поверхностное натяжение в лиофильных дисперсных системах в зависимости от размеров частиц может иметь значение от 1,410-7 до 1,410-3 Дж/м2. Типичными представителями лиофильных дисперсных систем являются растворы коллоидных поверхностно активных веществ (ПАВ) (ассоциативные коллоиды) и растворы полимеров (молекулярные коллоиды). Лиофобные системы термодинамически неустойчивы, т.к. частицы дисперсной фазы склонны к агрегации. Их агрегативная термодинамическая неустойчивость обусловлена избытком поверхностной энергии. Межфазное натяжение в них больше рассчитанного по соотношению Ребиндера – Щукина, поэтому они не могут быть получены самопроизвольным диспергированием. Для их образования должна быть затрачена внешняя энергия. Укрупнение частиц дисперсной фазы при потере агрегативной устойчивости достигается двумя путями:
В зависимости от природы среды и концентрации дисперсной фазы эти процессы могут заканчиваться или осаждением, или структурообразованием. При нарушении агрегативной устойчивости происходит коагуляция. Правила коагуляции электролитами. Порог коагуляции. Правило Шульце-Гарди. Виды коагуляции: концентрационная и нейтрализационная. Коагуляция смесями электролитов. Явление «неправильные ряды». Механизм и кинетика коагуляции Коагуляцией называется процесс слипания частиц с образованием крупных агрегатов. В результате коагуляции система теряет свою седиментационную устойчивость, так как частицы становятся слишком крупными и не могут участвовать в броуновском движении. Коагуляция является самопроизвольным процессом, так как она приводит к уменьшению межфазной поверхности и, следовательно, к уменьшению свободной поверхностной энергии. Различают две стадии коагуляции. 1 стадия – скрытая коагуляция. На этой стадии частицы укрупняются, но еще не теряют своей седиментационной устойчивости. 2 стадия - явная коагуляция. На этой стадии частицы теряют свою седиментационную устойчивость. Если плотность частиц больше плотности дисперсионной среды, образуется осадок. Причины коагуляции многообразны. Едва ли существует какое либо внешнее воздействие, которое при достаточной интенсивности не вызывало бы коагуляцию. |