Аспирин. Механизм действия аск представлен на
 Скачать 116.93 Kb. Скачать 116.93 Kb.
|

 Механизм действия АСК представлен на рисунке. Аспирин в первую очередь ингибирует ЦОГ тромбоцитов и эндотелия сосудов, подавляет образование тромбоксана А2 (мощного индуктора агрегации тромбоцитов и вазоконстрикции) — продукта метаболизма арахидоновой кислоты, а в больших дозах — синтез простациклина (дезагрегант и вазодилататор). АСК и другие нестероидные противовоспалительные препараты (индометацин) действуют на уровне метаболизма арахидоновой кислоты, а в больших дозах (Аспирин — от 300 до 500 мг) одновременно блокируют синтез простациклина, что косвенно может поддерживать агрегационное состояние тромбоцитов в результате количественного уменьшения простациклина и его дезагрегационного эффекта. В настоящее время выделяют четыре группы антитромбоцитарных препаратов, в основе разделения которых лежат принципы доказательной медицины, учитывающие эффективность и наличие побочных действий. Первая группа антитромбоцитарных препаратов, применение которых не рекомендуется для практической кардиологии вследствие отсутствия доказательной основы преимуществ перед Аспирином, неэффективности и потенциальной опасности: сульфинпиразон, дипиридамол, простациклин, блокаторы синтетазы тромбоксана А2, антагонисты рецепторов тромбоксана А2, препараты ингибиторов рецепторов IIb/IIIa тромбоцитов для приема внутрь. Вторая группа, составляющая основу современной антитромбоцитарной терапии, — ингибиторы циклооксигеназы (ЦОГ): ацетилсалициловая кислота (АСК) — Аспирин. Третья группа — тиенопиридины (клопидогрел — Плавикс, тиклопидин — Тиклин). Четвертая группа — блокаторы ГП-рецепторов IIb/IIIa для внутривенного применения (абциксимаб, эптифибатид, тирофибан, фрамон). Ацетилсалициловая кислота используется под торговым наименованием Аспирин с 1899 г. Хотя ацетилсалициловая кислота подвергалась интенсивным фармакологическим и клиническим исследованиям с самого момента её введения в лечебную практику, прошло почти 50 лет, прежде чем появились первые публикации, доказывающие влияние АСК на коагуляцию. Singer (1945) сообщил о поздних кровотечениях после тонзиллэктомии 3 у больных, принимавших АСК для ослабления послеоперационной боли. В последующие годы появлялись публикации, подтверждающие, что употребление АСК может вызвать изменения процесса гемостаза (Smith и Mackinnon, 1951; Wising, 1952). 2.2 ПОДАВЛЕНИЕ АГРЕГАЦИИ ТРОМБОЦИТОВ Только к концу 60-х годов было открыто (Weiss и Aledort, 1967) и затем систематически исследовалось (O’Brien, 1968 a, б; Weiss и соавт., 1968) ингибирующее действие АСК на функцию тромбоцитов. На основе собственных результатов O’Brien (1968) рекомендовал использовать АСК как антитромбоцитарный агент и определил дневную дозу 175 мг как достаточную для этой цели. Однако в то время механизм подавления функций тромбоцитов ацетилсалициловой кислотой был практически неизвестен. Только 3 года спустя Vane с сотрудниками (1971) разработали гипотезу, обеспечившую прорыв в понимании механизма действия ацетилсалициловой кислоты. Они предположили, что АСК и аналогичные родственные вещества ингибируют синтез простагландинов. Хотя Vane вначале описал ингибирование синтеза простагландинов только как механизм, объясняющий противовоспалительное действие АСК, этот подход обеспечил в первое время фармакологически приемлемое объяснение множественных клинических эффектов этой группы веществ. Smith и Willis в том же году показали, что АСК ингибирует синтез простагландинов в тромбоцитах, таким образом объяснив ее антитромбоцитарное действие. В ходе активного исследования простагландинов было выяснено, что АСК избирательно ингибирует циклооксигеназу (PGH синтазу4)- широко распространенный, связанный с мембранами фермент, действующий как катализатор биосинтеза простагландинов и других эйкозаноидов. Этот фермент присутствует по меньшей мере в двух изоферментных формах (Vane, 1994). С помощью рентгено-структурного анализа была определена структура одной из этих форм (Picot и соавт., 1994). Обобщающий термин “эйкозаноиды” обозначает группу биологически высокоактивных липидных медиаторов, содержащих 20 атомов углерода, что характерно для всех веществ этого класса (по гречески “эйкос” значит двадцать). Это обусловлено тем, что все эти вещества являются производными арахидоновой кислоты (20-углеродной полиненасыщенной жирной кислоты с четырьмя двойными связями), которая является составной частью фосфолипидов клеточной мембраны (Smith и соавт., 1991). После стимуляции клетки, например повреждением, арахидоновая кислота высвобождается из мембранных фосфолипидов и превращается в результате последовательности ферментных реакций (каскад арахидоновой кислоты) во множество различных метаболитов по двум основным путям – липоксигеназному и циклооксигеназному. Лейкотриены являются основными продуктами липоксигеназного метаболического пути, тогда как продуктами циклооксигеназного пути являются широко распространённые простагландины, включающие также простациклин, образующийся в эндотелии сосудов, и тромбоксан, который синтезируется в тромбоцитах (рис. 1). Сегодня известно более 150 эйкозаноидов, которые инициируют и/или модулируют различные регуляторные функции в организме. Хотя ацетилсалициловая кислота не влияет на липоксигеназу в изолированных человеческих тромбоцитах (оказывая тем не менее такое действие в плазме, обогащённой тромбоцитами), она является специфическим мощным ингибитором циклооксигеназы (см. обзор SchrЪr, 1992). Рис. 1. Упрощенная схема метаболизма арахидоновой кислоты по циклооксигеназному и липоксигеназному путям, показывающая блокаду циклооксигеназного пути ацетилсалициловой кислотой. 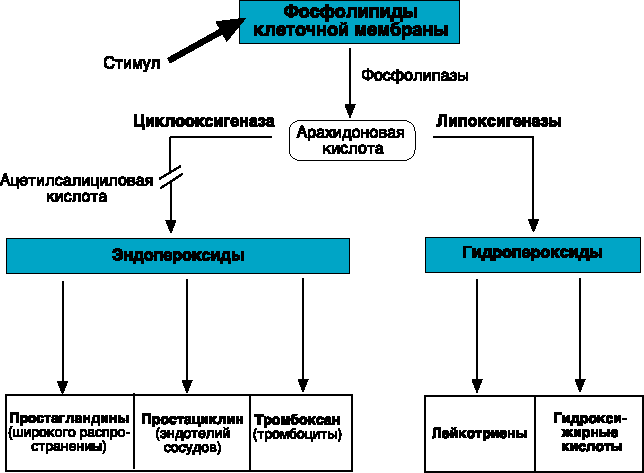 2.3 МЕХАНИЗМ ИНГИБИРОВАНИЯ ЦИКЛООКСИГЕНАЗЫ Через 4 года после открытия того факта, что АСК ингибирует синтез простагландина в тромбоцитах человека (Smith и Willis, 1971), Roth и Majerus (1975) смогли объяснить механизм этого явления. Они показали, что АСК ацетилирует циклооксигеназу тромбоцитов, тем самым необратимо блокируя ее. В ходе этого процесса ацетатная группа АСК ковалентно связывается с гидроксильной группой остатка серина на N-конце циклооксигеназы (Roth и Siok, 1978). Поэтому авторы сделали заключение, что эта аминокислота в активном участке молекулы фермента отвечает за его каталитическую активность и её блокада инактивирует весь фермент. Однако результаты более поздних молекулярных биохимических исследований привели к необходимости некоторого изменения этой гипотезы. Использование циклооксигеназы, полученной из семенных пузырьков барана (DeWitt и соавт., 1990), и клонированного фермента из человеческих тромбоцитов (Funk и соавт., 1991) дало возможность прийти к заключению, что АСК необратимо ацетилирует гидроксильную группу остатка серина в положении 529 (семенные пузырьки барана) или остатка серина в положении 530 (человеческие тромбоциты) в молекуле циклооксигеназы. Было также показано, что в отличие от предыдущего предположения этот сериновый остаток не является единственно возможным вариантом и может быть заменён другими подходящими аминокислотами без потери циклооксигеназной активности. Таким образом, в соответствии с современной концепцией, ацетилирование серинового остатка связано с неспецифическим стерическим ингибированием взаимодействия фермент-субстрат, ведущим к блокированию доступа арахидоновой кислоты к каталитическому центру циклооксигеназы (Smith с соавт., 1991; Shimokawa и Smith, 1992; рис. 2). Рис. 2. Молекулярный механизм ингибирования циклооксигеназы ацетилсалициловой кислотой. Ацетилирование остатка серина в положении 530 в молекуле циклооксигеназы в человеческих тромбоцитах приводит к ингибированию взаимодействия между этим ферментом и его субстратом арахидоновой кислотой (по Shimikawa и Smith , 1992). 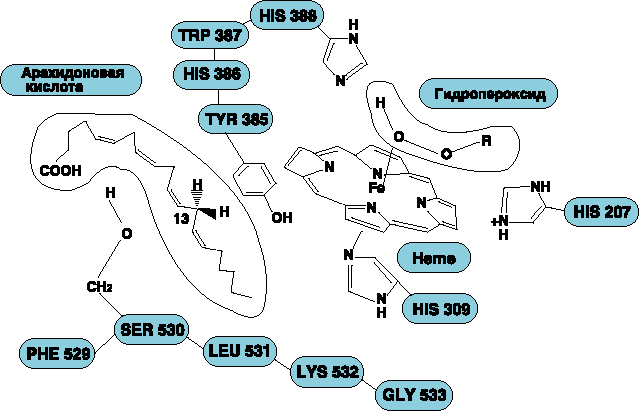 Описанные результаты теоретически обосновывают использование ацетилсалициловой кислоты для профилактики инфаркта миокарда, инсульта и тромбоэмболических осложнений после хирургических операций на кровеносных сосудах. Ингибирование циклооксигеназы в тромбоцитах в клиническом отношении является важным механизмом антикоагуляционного действия АСК (Schrцr, 1992). 2.4 ТРОМБОКСАН И ПРОСТАЦИКЛИН При каталитическом посредничестве циклооксигеназы арахидоновая кислота, высвободившаяся из мембранных фосфолипидов, превращается путём включения кислорода (оксигенация) и замыкания в кольцо (циклизация) в простагландин гидроэндопероксид PGG2. За этой реакцией циклооксигенации (оксигенация плюс циклизация) следует пероксидазная реакция, в которой пероксидаза восстанавливает PGG2 до соответствующего спирта PGH2. Оба эндопероксида химически нестабильны (рис. 3). Активность циклооксигеназы и пероксидазы является ферментной функцией комплекса PGH-синтазы. Ацетилсалициловая кислота ингибирует только реакцию циклооксигенации и не влияет на пероксидазную реакцию (SchrЪr, 1992). Это только предположение, так как специфичные для клеток изоферментные варианты циклооксигеназы и PGH-синтаза (Xie и соавт., 1991, 1992; Vane, 1994), а также тромбоксан А2 (TXA2) образуются из эндопероксидов простагландинов в тромбоцитах. TXA2 обладает сосудосуживающими свойствами и способствует агрегации тромбоцитов (Hamberg и соавт., 1975). Основное действие TXA2 выражается во второй фазе агрегации тромбоцитов, которая характеризуется высвобождением медиаторов тромбоцитов (таких, как серотонин, аденозин дифосфат и различные тромбоцитарные факторы). Это делает возможным слипание и агрегацию тромбоцитов, а затем их аутокаталитическую активацию (Coleman и соавт., 1987; Reilly и FitzGerald, 1988). Кроме того, TXA2 за счет инициации тромбов способствует развитию атеросклеротических бляшек и увеличению размеров поражения, усиливаемого PDGF (тромбоцитарный фактор роста) (Fitzgerald и соавт., 1983 a; SchrЪr, 1991). Рис. 3. Синтез нестабильных эндопероксидов простагландинов PGG2 и PGH2, а также стабильных простагландинов и тромбоксана А2 в циклооксигеназном пути метаболизма арахидоновой кислоты. Ацетилсалициловая кислота выборочно и необратимо ингибирует циклооксигеназу путем ацетилирования фермента, при этом сама превращается в салициловую кислоту (с изменениями по SchrЪr, 1992). 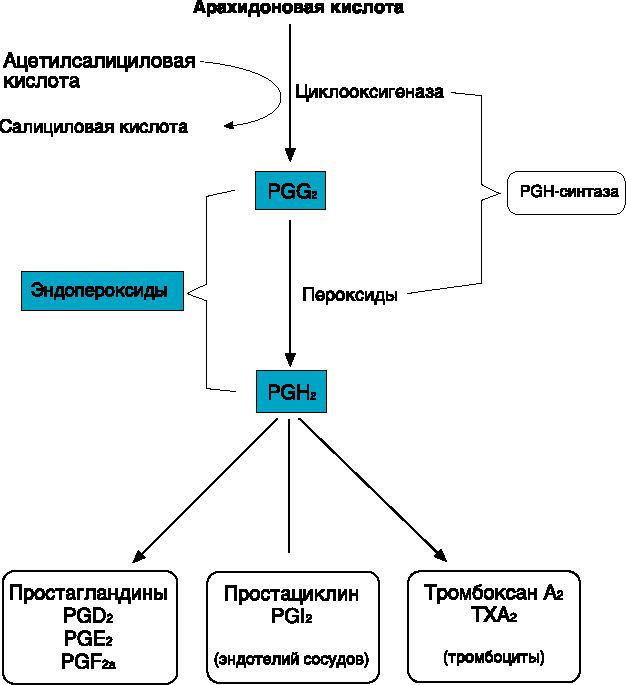 TXA2 может также играть роль медиатора ишемии, которая связана с агрегацией тромбоцитов. Поэтому образцы крови из коронарного синуса, взятые у больных, страдающих коронарной болезнью сердца, через короткий промежуток времени после приступа стенокардии, содержат повышенные концентрации TXB2, стабильного метаболита TXA2 (Hirsh и соавт., 1981; Robertson и соавт., 1981). Тромбоксан A2 нестабилен и характеризуется коротким временем полужизни в плазме крови – примерно 30 с. Таким образом, его действие, по-видимому, имеет преимущественно местный характер (Oates и соавт., 1988 a, 1988 б). Клетки эндотелия сосудов синтезируют большое количество простациклина (PGI2) и очень мало тромбоксана (Sinzinger и Firbas, 1983). В отличие от тромбоксана простациклин подавляет агрегацию тромбоцитов и вызывает расширение сосудов (Moncada и соавт., 1976; Moncada и Vane, 1979 a). Клиническое применение ацетилсалициловой кислоты при сердечно-сосудистых расстройствах определяется преимущественно этим прямым физиологическим антагонизмом между тромбоксаном и простациклином (Moncada и Vane, 1979 b; Mitchell, 1981; Gross, 1992; рис. 4). Рис. 4. Физиологический антагонизм между простациклином (PGI 2) из эндотелия кровеносных сосудов и тромбоксаном А2 (TXA2) из тромбоцитов. 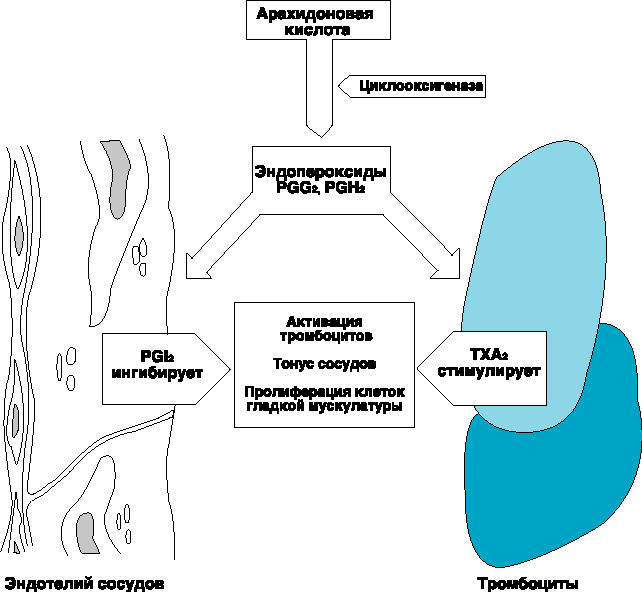 2.5 ПОСЛЕДСТВИЯ ИНГИБИРОВАНИЯ ЦИКЛООКСИГЕНАЗЫ Ацетилсалициловая кислота подавляет как синтез тромбоксана в тромбоцитах, так и синтез простациклина в клетках эндотелия кровеносных сосудов, однако в разной степени и с различными последствиями. С одной стороны, АСК более эффективно ингибирует синтез тромбоксана, чем синтез простациклина, потому что циклооксигеназа тромбоцитов, по-видимому, более чувствительна к ингибирующему воздействию АСК, чем фермент клеток эндотелия (Burch и соавт., 1978; Villa и соавт., 1979; Hanley и соавт., 1981; Preston и соавт., 1981; Wu и соавт., 1981; Weksler и соавт., 1985; Cerletti и соавт., 1987; Ritter и соавт., 1989). Однако фундаментальное значение имеет тот факт, что безъядерные тромбоциты теряют способность синтезировать белки и, таким образом, не могут заменять необратимо подавляемую циклооксигеназу путём компенсационного ресинтеза. Это означает, что они более не способны продуцировать тромбоксан в течение всего оставшегося времени их существования, которое в целом составляет приблизительно 10 дней. Только новообразующиеся тромбоциты являются функционально полноценными (Burch и соавт., 1983; Bye и соавт., 1979; Moncada и Vane, 1979 б; Boysen и соавт., 1982; Di Minno и соавт., 1983; FitzGerald и соавт., 1983 б; Whitte и Moncada, 1983; Patrono, 1984; Patrono и Patrignani, 1984). Таким образом, синтез тромбоксана человеческими тромбоцитами остаётся подавленным примерно на 90% в течение 3 дней после однократного перорального приёма 200 мг АСК (Patrono и соавт., 1980). После употребления последней дозы АСК проходит в среднем 7 –10 дней до нормализации содержания тромбоксана, измеренного по содержанию его стабильного метаболита TBX2. Это восстановление происходит только за счет образования в костном мозге новых тромбоцитов (Reilly и FitzGerald, 1988; Sinzinger и соавт., 1988). Длительное действие АСК на функцию тромбоцитов объясняется необратимым ингибированием циклооксигеназы, которая “отличает” АСК от других антикоагуляционных агентов, обладающих обратимым действием, длящимся лишь несколько часов. После приема АСК подавление агрегации тромбоцитов происходит довольно быстро. Скорость наступления действия зависит прежде всего от скорости гидролиза АСК (Akopov и соавт., 1992). При употреблении препаратов АСК (ЭС) максимальное подавляющее действие достигается через 3-4 часа. Для ускорения действия АСК таблетки следует перед проглатыванием разжевать. При этом максимальное ингибирование функций тромбоцитов достигается через 15-30 мин. (Jimenez и соавт., 1989). Клетки эндотелия кровеносных сосудов, имеющие в отличие от тромбоцитов ядра, способны ресинтезировать циклооксигеназу, инактивированную АСК. Поэтому недостаток простациклина полностью и быстро компенсируется. Таким образом, подавляющее действие АСК на эндотелиальные клетки менее продолжительно, чем соответствующее действие на тромбоциты (Masotti и соавт., 1979; Jaffe и Weksler, 1979; Buchanan с соавт., 1980). Даже при употреблении высоких доз АСК, например 2 раза в сутки по 500 мг (Vesterquist, 1986), 600 мг (Heavey и соавт., 1985) и 600 мг в виде болюсной внутривенной инъекции плюс 600 мг/ч внутривенно путём 3-часового вливания (Ritter и соавт., 1989), активность циклооксигеназы полностью восстанавливается в течение 3-6 ч после приема дозы препарата. 2.6 ДОЗОЗАВИСИМОСТЬ ИНГИБИРОВАНИЯ ЦИКЛООКСИГЕНАЗЫ Patrono (1989) с целью уточнения дозировки АСК, необходимой для ингибирования циклооксигеназы тромбоцитов у здоровых людей, исследовал результаты однократного и повторного приема различных ее доз. Он обнаружил, что для 50% ингибирования синтеза тромбоксана (ID50) доза АСК составляет 26 мг при однократном приеме и 3.2 мг в случае употребления в несколько приёмов в течение 5 дней. Отношение 1:8, соответствующее этим дозам, практически точно отражает долю новообразующихся тромбоцитов, поступающих ежедневно в кровоток, в общем количестве тромбоцитов в периферической крови. При непрерывной терапии только новообразующаяся доля тромбоцитов, составляющая 1/8 от их общего числа, нуждается в ацетилировании, поэтому для поддержания терапевтического действия достаточно такой небольшой дозировки (3.2 мг в сутки). Основываясь на этих результатах, Patrono вычислил, что при непрерывной терапии доза, составляющая около 40 мг АСК в сутки, достаточна для полного ингибирования синтеза тромбоксана. Перекрёстное сравнение дозировок 10 и 30 мг АСК в сутки в плацебо-контролируемом, двойном слепом исследовании на здоровых добровольцах показало, что при употреблении 30 мг АСК в сутки в течение 3 недель синтез тромбоксана в тромбоцитах снижается на 94% и время кровотечения увеличивается относительно контрольной величины до 160%. Однако при этом общий уровень синтеза простациклина в организме, который измерялся по выведению с мочой 6-оксо-PGF1a, оставался постоянным (Kallmann и соавт., 1987). При многократном употреблении доз 20 и 50 мг АСК уменьшение синтеза тромбоксана становилось заметным через 2-4 дня. Через 3 недели эта величина достигала 10% от начального уровня (Sinzinger и соавт., 1988; 1989). Серией исследований было доказано, что АСК в суточной дозе 0.5-1 мг/кг или 30-70 мг полностью ингибирует синтез тромбоксана в тромбоцитах у здоровых людей в течение одной недели, не оказывая при этом значительного влияния на общий уровень образования простациклина или PGE2 в клетках сосудов (Patrignani и соавт., 1982; FitzGerald и соавт., 1983 а; Zaragoza и Breton, 1987; Lorennz и соавт., 1989; Vanags и соавт., 1990; Chiabrando и соавт., 1992). Однако в более поздних исследованиях было показано, что, несмотря на значительное изменение общего уровня синтеза тромбоксана, происходит существенное локальное ингибирование PGI2 даже при употреблении АСК в дозе 20-40 мг в сутки (Cerletti и соавт., 1987; Chiabrando и соавт., 1992). Так, АСК в дозе 40 мг в течение 4 дней уменьшает синтез простациклина у здоровых людей по данным биопсии вен более чем на 80% (Preston и соавт., 1981). В плацебо-контролируемом, двойном слепом перекрёстном исследовании, проведённом Kyrle и сотр. (1987 a, б), после применения 35 мг АСК в сутки в течение 7 дней было обнаружено снижение синтеза тромбоксана и простациклина на 80 – 90% в месте образования тромба после повреждения сосуда. Несмотря на сниженный синтез PGI2, в обоих исследованиях наблюдалось значительное увеличение времени кровотечения, что подтверждается также исследованиями Wersler и соавт. (1985). Полученные результаты свидетельствуют о том, что увеличение продолжительности кровотечения после употребления АСК, по- видимому, вызвано прежде всего уменьшением синтеза тромбоксана, а не снижением образования простациклина. Такие же низкие дозы АСК достаточны для ингибирования синтеза тромбоксана и у больных стабильной стенокардией. Современные данные не позволяют высказаться с определенностью, обладают ли тромбоциты больных нестабильной стенокардией или острым инфарктом миокарда пониженной чувствительностью к малым дозам АСК (40 мг в сутки) (De Caterina и соавт., 1985; Rasmanis с соавт., 1988; Noseda и соавт., 1989). Неясно также, повышает ли АСК уменьшенную чувствительность тромбоцитов к простациклину у больных острым инфарктом миокарда (Gasser и соавт., 1990). 2.7 ИНГИБИРОВАНИЕ ЦИКЛООКСИГЕНАЗЫ У БОЛЬНЫХ С СОСУДИСТЫМИ НАРУШЕНИЯМИ При сосудистых поражениях синтез простациклина нарушается из-за дисфункции эндотелия. Это приводит к относительной недостаточности PGI2 и преобладанию TXA2 при общей тенденции к гиперреактивности тромбоцитов, которая становится особенно заметной при напряжении системы. До сих пор не было убедительных данных о небходимости применения более высоких доз АСК (> 50 мг в сутки) у больных с заболеваниями сосудов для ингибирования синтеза тромбоксана и функции тромбоцитов по сравнению со здоровыми людьми (FitzGerald и соавт., 1985; Boysen и соавт., 1988; De Caterina и соавт., 1990). Несмотря на то, что у больных с дегенеративными заболеваниями кровеносных сосудов АСК ингибирует синтез простациклина в стенке сосудов в большей степени, чем у здоровых людей, из этого необязательно следует, что у таких больных циклооксигеназа эндотелия более чувствительна к действию АСК. Альтернативное объяснение может состоять в том, что клетки эндотелия с нарушенной функцией не образуют достаточного количества предшественников простагландина (PG-endoperoxides) для внутреннего синтеза PGI2 из-за относительного дефицита циклооксигеназы, который может компенсироваться поступлением повышенного количества эндопероксидов из гиперреактивных тромбоцитов, прилипших к стенкам сосуда (Dewitt и Smith, 1983). В этой модели эндотелиальный синтез простациклина более зависим от поступления эндопероксидов из тромбоцитов. В связи с этим ингибирование ацетилсалициловой кислотой тромбоцитарной циклооксигеназы должно не только снижать продукцию тромбоцитами эндопероксидов и тромбоксана, но и подавлять синтез простациклина в сосудистой стенке вследствие снижения поступления эндопероксидов из тромбоцитов. Согласно этой концепции, ингибирующее действие будет тем сильнее, чем более эндотелиальный синтез простациклина зависит от эндопероксидов, поступающих из тромбоцитов (SchrЪr, 1992). Эта гипотеза подтверждается наблюдением, что эндотелиальные клетки in vitro поглощают PG-endoperoxides, образовавшиеся в тромбоцитах, что и делает возможным синтез простациклина (Marcus и соавт., 1980). Результаты исследований in vivo также показали, что производство простациклина в эндотелии повышается в месте поражения, как только тромбоциты начинают налипать на стенки сосуда (Nowak и FitzGerald, 1989). Эта концепция подтверждается также тем фактом, что ингибиторы синтеза тромбоксана усиливают действие простагландинов в присутствии активированных тромбоцитов (Mullane и Fornabaio, 1988), а также тем, что у больных, страдающих тяжёлым атеросклерозом и внутрисосудистой активацией тромбоцитов, синтез и выведение метаболитов простациклина повышены и тесно коррелируют друг с другом (FitzGerald и соавт., 1984). С учетом этих результатов можно предположить, что при дисфункции эндотелия синтез простациклина в клетках сосудов повышен из-за усиленного поступления предшественников простагландина из налипших на стенки сосуда тромбоцитов (SchrЪr, 1992). У больных с заболеваниями сосудов в отличие от здоровых невозможно выявить отчётливую связь между действием низких доз АСК на простациклин и синтезом тромбоксана. Поэтому у больных, подвергшихся шунтированию, достаточно однократного приема АСК в дозе 40 мг для ингибирования образования простациклина (по измерениям в биоптатах сосудов) (Weksler и соавт., 1983). У больных с атеросклерозом употребление АСК по 20 мг в сутки в течение одной недели вызывало ингибирование синтеза PGI2 в аорте и венах приблизительно на 50% (Weksler и соавт., 1985). У пожилых больных с далеко зашедшим окклюзирующим поражением выведение почками метаболитов как тромбоксана, так и простациклина заметно снижалось после употребления АСК по 50 мг в сутки в течение недели (Carlsson и соавт.,1990). Tsang с соавт. (1988) обнаружили, что у больных после коронарного шунтирования и длительного перорального приёма АСК по 75-100 мг в сутки синтез простациклина в аорте снизился на 95%. Knapp и сотр. (1988) также не смогли продемонстрировать отчётливое различие действия низких доз АСК (по 20 мг 2 раза в сутки) на тромбоциты и кровеносные сосуды у пожилых больных с сосудистыми нарушениями в отличие от здоровых людей. У больных, страдающих атеросклерозом, выведение метаболитов тромбоксана и простациклина почками было соответственно в 5 и 2.5 раза выше, чем у здоровых людей. Возраст больных не оказывал существенного влияния на эти показатели. Выведение метаболитов тромбоксана после приема АСК снижалось одинаково во всех группах (на 80%). С другой стороны, ацетилсалициловая кислота в 2 раза сильнее подавляла выведение метаболитов простациклина у больных с атеросклерозом, чем у здоровых людей (рис. 5). Рис. 5. Почечная экскреция метаболитов тромбоксана и простациклина у молодых здоровых людей (контроль 1), у больных с атеросклерозом и здоровых людей того же возраста (контроль 2) до и после семидневного курса лечения ацетилсалициловой кислотой в дозе 20 мг два раза в сутки. Подробности см. в тексте (по Knapp с соавт., 1988). 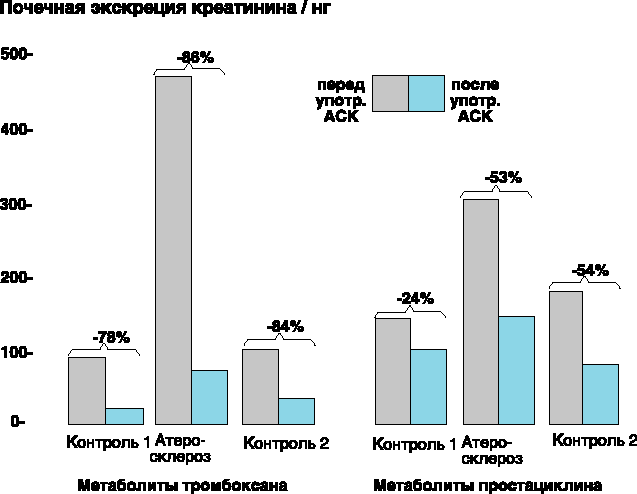 Эти результаты, по-видимому, поддерживают концепцию, согласно которой снижение выведения простациклина после употребления АСК у больных , страдающих тяжёлым атеросклерозом, происходит прежде всего из-за подавления функций тромбоцитов, а не вследствие действия, оказываемого на клетки эндотелия сосудов (FitzGerald и соавт., 1985). Придерживаясь этой точки зрения, Force и сотр. (1991) показали, что у больных с инфарктом миокарда ингибирование синтеза простациклина ацетилсалициловой кислотой происходит в результате уменьшения поступления метаболических предшественников из тромбоцитов. Если эта гипотеза верна, то низкие дозы АСК должны снижать синтез простациклина в клетках эндотелия сосудов пропорционально дефициту предшественников, поставляемых тромбоцитами. 2.8 ГЕМОСТАЗ И ТРОМБОГЕНЕЗ В физиологических условиях повреждение кровеносного сосуда инициирует в течение нескольких секунд сложные и мощные механизмы остановки кровотечения для защиты организма от опасных для жизни потерь крови. Основную роль в инициации этих процессов играет взаимодействие между тромбоцитами и стенкой сосуда (Marcus и Safier, 1993). Повреждение эндотелия кровеносного сосуда (например, механическое, токсическое или при локальных нарушениях кровотока) инициирует процесс гемостаза (Gertz и соавт., 1981). В сосудах, пораженных атеросклерозом, наиболее часто местом адгезии тромбоцитов к стенке являются изъязвленные бляшки или бляшки с щелевидными надрывами, обеспечивающие контакт пластинок с богатыми коллагеном субэндотелиальными структурами (Davies и Thomas, 1981; Ross, 1988). Экспериментально удалось показать, что атеросклеротические изменения сосудов никогда не развиваются без предварительного повреждения эндотелия и последующей агрегации в этом месте тромбоцитов (Harker и соавт., 1981; Ross, 1993). “Прилипание” тромбоцитов к соединительной ткани субэндотелия осуществляется посредством специальных связывающих белков (фактор фон Виллебранда, фибронектин, мембранные гликопротеины Ia и Ib), которые высвобождаются тромбоцитами и субэндотелиальными клетками и связывают взаимодействующие структуры (Siess, 1989). Коллаген и некоторые плазматические факторы, такие, как катехоламины, аденозин дифосфат и тромбин, “активируют” тромбоциты, что можно распознать по изменению их формы. В ходе сферической трансформации тромбоциты сначала теряют свою нормальную форму диска и принимают выпукло-вогнутые и сферические формы (Gerrard, 1988; рис. 6). Активируется метаболизм арахидоновой кислоты (при этом медиатором является тромбоксан А2), из тромбоцитов высвобождаются такие вещества, как АДФ5, серотонин и различные тромбоцитарные факторы (“реакция высвобождения”). Это приводит к активации других тромбоцитов, что значительно усиливает реакцию (Baumgartner и соавт., 1976; Cazevane, 1977; Coleman c соавт., 1987; Reilly и FitzGerald, 1988). Активацию тромбоцитов можно распознать морфологически по образованию псевдоподий и длинных выростов поверхностной мембраны (Scharf и соавт., 1983). Агрегация тромбоцитов представляет собой образование сетевидных или бугорчатых агломератов тромбоцитов путём связывания фибриногена со специфическими рецепторами на их поверхностной мембране (гликопротеиновый комплекс IIb-IIIa; Gerrard, 1988). Вместе с высвобождением сосудосуживающих веществ из гранул тромбоцитов (серотонин) этот процесс способствует остановке кровотечения (образование гемостатической пробки и сужение сосуда). Рис. 6. Начальная стадия тромбогенеза: трансформация, образование псевдоподий, прилипание и агрегация тромбоцитов.  Вторая фаза гемостаза характеризуется активацией свертывающей системы плазмы тромбоцитарным фактором 3 и тромбопластином (тромбокиназа), поступающими из повреждённой ткани. Этот процесс коагуляции, оканчивающийся превращением фибриногена плазмы в фибрин, стимулирует дальнейшие адгезию и агрегацию тромбоцитов. Тромбоцитарный фактор 4 ингибирует гепарин, уменьшая таким образом фибринолитическую активность. Начальным результатом реализации этих механизмов является образование в стенке сосуда микротромба, который в ходе дальнейшего тромбогенеза развивается в механически прочный тромб путем присоединения новых агрегатов тромбоцитов и эритроцитов, вместе с образованием сети нитей фибрина между активированными тромбоцитами. Таким образом дефект в стенке сосуда закупоривается. Из-за уменьшения кровотока в месте стаза при наличии атеросклеротических бляшек этот процесс может привести к полной закупорке сосуда. В нормальных условиях образование тромба ограничено местом повреждения стенки сосуда, так как одновременно в прилегающих неповреждённых участках эндотелия активируются механизмы, подавляющие функции тромбоцитов и вызывающие расширение сосуда (образование простациклина, оксида азота, аденозина), что противодействует избыточной коагуляции. Возможно, агрегация тромбоцитов в месте повреждения стенки сосуда является “пусковым” фактором (Nowak и FitzGerald, 1989). 2.9 ПАТОФИЗИОЛОГИЧЕСКИЕ АСПЕКТЫ ГЕМОСТАЗА Неотъемлемой чертой гомеостаза системы коагуляции является динамический баланс, существующий в физиологических условиях между факторами, поступающими из сосудистой стенки (в частности, простациклин, вырабатываемый клетками эндотелия сосудов), и факторами, образующимися в циркулирующих клетках крови (в частности, тромбоцитарный тромбоксан). При таких дегенеративных заболеваниях сосудов, как выраженный атеросклероз или окклюзирующее поражение периферических артерий, это равновесие нарушается. Особенно нарушается синтез и высвобождение таких эндотелиальных медиаторов, как простациклин, оксид азота и тканевый активатор плазминогена (t-PA). Это связано с нарушением регуляторных функций эндотелия. Однако существуют признаки нарушения также и тромбопоэза, происходящего в костном мозге (Martin и соавт., 1991). Эффект гиперреактивности циркулирующих тромбоцитов клинически соответствует состоянию гиперкоагуляции. Дегенеративные заболевания сосудов сопровождаются вазоспазмом, обусловленным TXA2 и серотонином, а также катехоламинами (Evans и соавт., 1968; Schrцr и Braun, 1990). Это имеет большое значение для тромбогенеза, так как локальный вазоспазм может вызывать тромбоэмболическую окклюзию при наличии предсуществующего стеноза сосудов (Santamore и соавт., 1991). Так как эндотелий производит фибринолитические регуляторные факторы (t-PA), фибринолиз при дегенеративных заболеваниях сосудов подавляется, что способствует повышению тенденции к тромбозам. Ацетилсалициловая кислота оказывает влияние на все три составные части коагуляции: функции тромбоцитов, плазматическую коагуляцию и фибринолиз. 2.10 ДЕЙСТВИЕ АЦЕТИСАЛИЦИЛОВОЙ КИСЛОТЫ НА ФУНКЦИИ ТРОМБОЦИТОВ Механизмом, лежащим в основе антитромбоцитарного действия АСК, является ингибирование синтеза тромбоксана. In vitro оно достигается максимум через 15 мин. (Zuker и Peterson, 1970). Клинически значимое действие, о котором можно судить по увеличению времени кровотечения, достигается только при практически полном ингибировании образования тромбоксана (Reilly и FitzGerald, 1987). Однако сократимость цитоскелета тромбоцитов и, следовательно, первичная агрегация тромбоцитов при этом не затрагиваются (Pengo и соавт., 1985). То же самое можно сказать и про тромбинзависимую активацию тромбоцитов. Тромбоксан играет ограниченную роль в активации тромбоцитов, обусловленной АДФ и механическим стрессом (например, надрыв). Поэтому АСК мало влияет на эти механизмы (Moake и соавт., 1988; Yao и соавт., 1922; Rajagopalan и соавт., 1988; Ratnatunga и соавт., 1992). Кроме того, АСК не оказывает влияния на образование в эндотелии оксида азота и аденозина, которые, как и простациклин, подавляют функции тромбоцитов, противодействуя таким образом патологической активации тромбоцитов (Marcus и соавт., 1991). Интересно, что АСК может противодействовать острому повреждающему действию никотина на тромбоциты, эндотелий и липиды плазмы у курильщиков (Blanche и соавт., 1992). Потенциальная клиническая значимость этих результатов в отношении атерогенного влияния курения до сих пор остаётся неясной. Однако результаты больших проспективных исследований не подтверждают предположение, что АСК предотвращает прогрессирование атеросклероза (Manson и соавт., 1990; Ridker и соавт., 1991 а). 2.11 ДЕЙСТВИЕ АЦЕТИЛСАЛИЦИЛОВОЙ КИСЛОТЫ НА КОАГУЛЯЦИЮ По имеющимся наблюдениям, ацетилсалициловая кислота в дозах до 1 г в сутки не оказывает какого-либо действия на плазматическую коагуляцию (BjЪrnsson и соавт., 1989; Fiore и соавт., 1990). У больных гемофилией АСК увеличивает время кровотечения так же, как и у здоровых людей (Miehlke, 1981). Эти результаты подтверждают гипотезу, что увеличение времени кровотечения при лечении АСК объясняется прежде всего подавлением функций тромбоцитов. Более высокие дозы АСК (больше 3-4 г) подавляют плазматическую коагуляцию путём нарушения синтеза протромбина (Wising, 1952) и уменьшения образования факторов коагуляции VII, IX и X. Кроме того, АСК также ацетилирует антитромбин III, что может приводить к ослаблению действия гепарина (Villanueva и Allen, 1986). 2.12 ДЕЙСТВИЕ АСК НА ФИБРИНОЛИЗ Ацетилсалициловая кислота оказывает различное воздействие на фибринолиз. У здоровых людей АСК ингибирует усиление фибринолиза, вызванное ишемией. Это действие объясняется уменьшением высвобождения профибринолитического эндотелиального простациклина и t-PA. При этом активность ингибитора активатора плазминогена (PAI-I) не изменяется (Keber и соавт., 1987; Woods и Lazzari, 1987; Levin и соавт., 1989; Bertelй и соавт., 1989). Большее клиническое значение имеет тот факт, что АСК оказывает синергическое действие на фибринолитический эффект стрептокиназы (Basinski и соавт., 1991). Это объясняется подавлением АСК функции тромбоцитов, что противодействует их активации стрептокиназой и продуктами деградации фибрина (FitzGerald и соавт., 1988). В связи с прямой стимуляцией тромбоцитов стрептокиназой перед тромболизом рекомендуется ингибировать их функцию (Heptinstall и соавт., 1990). Кроме того, АСК стимулирует фибринолиз путём ацетилирования фибриногена, повышая, возможно, чувствительность фибрина в тромбе к лизису (BjЪrnsson и соавт., 1989). Клиническое значение синергизма между ацетилсалициловой кислотой и стрептокиназой было убедительно показано в исследовании ISIS-2 (1988). У здоровых людей АСК не влияет ни на исходную активность t-PA и PAI-I (Krishnamurti и соавт., BjЪrnsson и соавт., 1989), ни на синтез антиплазмина (Woods и Lazzari,1988), не усиливая фибринолиз после повреждения сосудов (Kyrle и соавт., 1987 a, б) и высокой физической активности (Keber и соавт., 1987) У больных с дисфункцией эндотелия действие АСК на фибринолиз отличается от такового у здоровых людей: у больных атеросклерозом с приступами преходящей ишемии было показано снижение исходной активности t-PA (Hampton и соавт., 1990). У больных с транзиторной ишемией стимуляция фибринолиза не может быть подавлена ацетилсалициловой кислотой (Carriero и соавт., 1987). |