Наследственные болезни и их классификация
 Скачать 2.04 Mb. Скачать 2.04 Mb.
|

1 2  НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ И ИХ КЛАССИФИКАЦИЯПроблема здоровья людей и генетика тесно взаимосвязаны. Ученые-генетики пытаются ответить на вопрос, почему одни люди подвержены различным заболеваниям, в то время как другие в этих же, или даже худших условиях остаются здоровы. В основном это связано с наследственностью каждого человека, т.е. свойствами его генов, заключенных в хромосомах. В последние годы отмечаются быстрые темпы развития генетики человека и медицинской генетики. Это объясняется многими причинами и прежде всего резким увеличением доли наследственной патологии в структуре заболевания и смертности населения. Статистика показывает, что из 1000 новорожденных у 35-40 выявляются различные типы наследственных болезней, а в смертности детей в возрасте до 5 лет хромосомные болезни составляют 2-3%, генные - 8-10%, мультифакториальные - 35-40%. Ежегодно в нашей стране рождается 180 тысяч детей с наследственными заболеваниями. Более половины из них имеют врожденные пороки, около 35 тысяч - хромосомные болезни и свыше 35 тысяч - генные болезни. Следует отметить, что число наследственных болезней у человека с каждым годом растет, отмечаются новые формы наследственной патологии. В 1956 году было известно 700 форм наследственных заболеваний, а к 1986 году их число увеличилось до 2000. В 1992 году количество наследственных болезней и признаков возросло до 5710. Все наследственные болезни делятся на три группы:
Генные болезни Генные болезни - это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена. Общая частота болезней в популяции составляет 1-2 %. Условно частоту генных болезней считают высокой, если она встречается с частотой 1 случай на 10000 новорожденных, средней - 1 на 10000 - 40000 и далее - низкой. Моногенные формы генных заболеваний наследуются в соответствии с законами Г.Менделя. По типу заболеваний они делятся на аутосомно-доминантные, аутосомно-рецессивные и сцепленные с Х- или Y-хромосомами. Большинство генных патологий обусловлено мутациями в структурных генах, осуществляющих свою функцию через синтез полипептидов - белков. Любая мутация гена ведет к изменению структуры или количества белка. Начало любой генной болезни связано с первичным дефектом мутантного аллеля. Основная схема генных болезней включает ряд звеньев: мутантный аллель→ измененный первичный продукт →цепь последующих биохимических процессов клетки → органы → организм.   В результате мутации гена на молекулярном уровне возможны следующие варианты: В результате мутации гена на молекулярном уровне возможны следующие варианты:
Особенностью генных (как и вообще всех наследственных) болезней является их гетерогенность. Это означает, что одно и то же фенотипическое проявление болезни может быть обусловлено мутациями в разных генах или разными мутациями внутри одного гена. Впервые гетерогенность наследственных болезней была выявлена С.Н.Давиденковым в 1934 г. К генным болезням у человека относятся многочисленные болезни обмена веществ. Они могут быть связаны с нарушением обмена углеводов, липидов, стероидов, пуринов и пиримидинов, билирубина, металлов и др. Пока еще нет единой классификации наследственных болезней обмена веществ. Научной группой ВОЗ предложена следующая классификация: 1) болезни аминокислотного обмена (фенилкетонурия,алкаптонурия); 2) наследственные нарушения обмен углеводов(галаюгоземия, гликогеновая болезнь и др.); 3) болезни, связанные с нарушением липидного обмена (болезнь Ниманна-Пика, болезнь Гоше и др.); 4)наследственные нарушения обмена стероидов; 5)наследственные болезни пуринового и пиримидинового обмена (подагра,синдром Леша-Найяна и др.); 6) болезни нарушения обмена соединительной ткани (болезнь Марфана, мукополисахаридозы и др.); 7)наследственные нарушения гемма- и порфирина(гемоглобинопатия);
9)наследственные нарушения обмена билирубина; 10) наследственные болезни обмена металлов (болезнь Коновалова-Вильсона и др.); 11) наследственные синдромы нарушения всасывания в пищеварительном тракте (муковисцидоз, непереносимость лактозы и др.). Рассмотрим наиболее часто встречающиеся и генетически более изученные в настоящее время генные болезни. Фенилкетонурия Причина: Генный дефект - отсутствие или недостаточная активность фермента фенилаланингидроксидазы, что приводит к нарушению обмена кислоты фенилаланина (содержание ее повышается до 20-60%). Это ведет к побочным путям превращения фенилаланина с образованием фенилкетонов, которые выделяются с мочой. Вторичное нарушение этой кислоты и триптофана приводит к нарушению синтеза необходимых для развития и жизнедеятельности нервных клеток веществ. Нарушается образование меланина, что приводит к недостаточной окраске кожных покровов. Тип наследования: Аутосомно-рецессивный. Клиника: При рождении ребенок выглядит совершенно нормальным. Примерно с 6 месяцев начинают проявляться клинические признаки заболевания. Возникают признаки поражения нервной системы: утрачиваются сформированные ранее нервно-психические навыки, наблюдается резкое отставание в развитии, психомоторное возбуждение с приступами неадекватных выкриков, смеха, стереотипные движения, судорожные припадки. У большинства детей светлые волосы и голубая радужная оболочка, на коже — экзема, уменьшение размеров черепа, деформация ушных раковин. Патогенез: Мышечное повышение тонуса, атаксии и гиперкинез. Врожденные пороки развития (сердца, костной системы). Диагностика: (крайне важна, так как лечебные мероприятия могут предотвратить развитие нервно-психических нарушений). Клиническое и биохимическое обследование мочи и крови: реакция мочи с хлоридом железа (III), при наличии заболевания появляется грязно-зеленое окрашивание; определение количества фенилаланина в крови. Лечение: Диета, продукты, лишенные фенилаланина, витаминные препараты. Алкаптонурия Причина: Нарушение обмена фенилаланина и тирозина и экскреция с мочой гомогентизиновои кислоты. Тип наследования: Аутосомно-рецессивный. Клиника: Заболевание у детей протекает без субъективных жалоб. Единственным симптомом является выделение темной мочи. Психофизическое развитие в норме. Патогенез: Отложение темного пигмента в хрящах, надпочечниках, щитовидной, поджелудочной, предстательной железах, деформация хрящей и суставов. Диагностика: Биохимическое обследование, по клиническим проявлениям: на пеленках, смоченных мочой, остаются темно-коричневые пятна. Лечение: Большие дозы аскорбиновой кислоты. Цистинурия Причина: Генный дефект, вызвавший дефицит фермента некоторых путей трансмембранного транспорта (цистатионинсинтетазы). Нарушение обмена цистатионинсинтетазы. Тип наследования: Аутосомно-рецессивный. Клиника: Симптомы заболевания проявляются спустя некоторое время после рождения ребенка. Снижен аппетит, отставание в росте и весе. Патогенез: Образование цистиновых камней в почках. Диагностика: Клиническое и биохимическое обследование. Лечение: Снижение количества потребляемого метионина. Диета включает большое количество цистинина и терапевтические дозы витамина В6. Катаракта Причина: Генная мутация, обусловливающая врожденное помутнение хрусталика. Тип наследования: Аутосомно-доминантный, реже аутосомно-рецессивный. Клиника: Снижение зрения от незначительного ослабления до полной потери светоощущения. Патогенез: Частичное или полное помутнение слоев хрусталика, сочетающееся с другими пороками развития глаз — нистагмом, косоглазием, микрофтальмией. Диагностика:Клиническое обследование. Лечение:Хирургическое устранение эффекта. Гемофилия Причина: Наследственный дефицит плазменного фактора свертывания крови в связи с прямой мутацией гена, локализованного в длинном плече X-половой хромосомы. Тип наследования: Рецессивный, сцепленный с Х-хромосомой. Клиника: На 1-м году жизни 1% кровотечений, с возрастом проявления более выражены. Дети, страдающие гемофилией, отличаются хрупкостью, бледной, тонкой кожей и слаборазвитым подкожным жировым слоем. Чрезмерные кровотечения при малейших повреждениях (гемотурия). В течение заболевания периоды кровоточивости сменяются периодами относительного благополучия. Больные - преимущественно мальчики. Патогенез: Подкожные и внутримышечные кровоизлияния и кровоизлияния во внутренние органы, поражение крупных суставов, что приводит к их деформации. Диагностика: Клиническое обследование и биохимический анализ крови, установление генеалогии. Лечение: Больным вливают антигемофильную плазму или криопрециптат (белковый концентрат с высокой коагуляционной активностью), назначают прямые трансфузии (не реже 3-х раз в сутки). Анемия Фанкони (наследственная апластическая) Причина: к настоящему времени выявлено восемь комплементационных групп при анемии Фанкони: FA-A, FA-B, FA-C, FA-D, FA-E, FA-F, FA-G, FA-H. Тип наследования: Аутосомно-рецессивный. Клиника: дефект развития лучевых костей и больших пальцев рук. Дети низкого роста, отмечается недоразвитие половых органов, микрофтальмия, косоглазие, коричневая пигментация кожи, почечная и сердечная недостаточность. Первые симптомы анемизации проявляются чаще от 6 месяцев до 4 лет. Длительность жизни детей не превышает 2-5 лет. Дети погибают от резкой анемизации, кровоизлияния в мозг или желудочно-кишечных кровотечений. Диагностика: по внешним признакам, клиническое обследование, биохимический анализ крови. Патогенез: повышенное содержание щелочноустойчивого гемоглобина, качественное изменение фермента гексокиназы, снижение образования АТФ, уменьшение пероксидазы в ядрах нормобластов костного мозга, понижение уровня щелочной фосфотазы и содержания фосфолипидов и полисахаридов.  Галактоземия ГалактоземияПричина: Происходит накопление в крови больного галактозы, что приводит к поражению многих органов: печени, нервной системы, глаз и др. Тип наследования: Аутосомно-рецессивный. Клиника: Болезнь проявляется с первых дней жизни расстройствами пищеварения, интоксикацией (понос, рвота, обезвоживание). У больных увеличивается печень. Обнаруживается катаракта (помутнение хрусталика глаза), умственная отсталость. Диагностика: Определение активности фермента галактоза-1-фос-фаттрансферазы в эритроцитах, а также галактозы в крови и моче, где уровни ее увеличены. Патогенез: У больных развивается печеночная недостаточность и желтуха. У погибших в первый год жизни детей при вскрытии обнаруживают цирроз печени. Лечение: При исключении из пищи молока (источника галактозы) и раннем назначении диеты больные дети могут нормально развиваться. 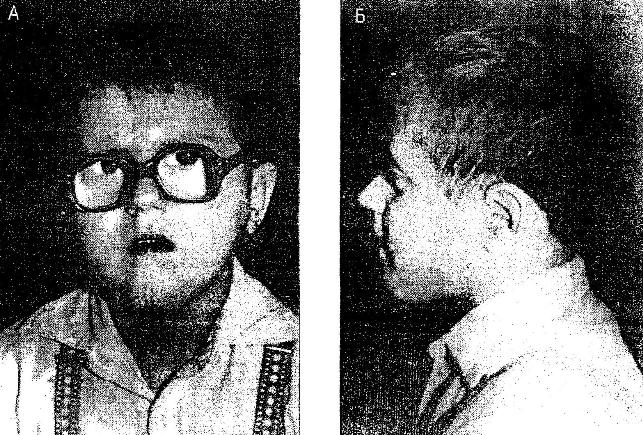 Болезнь Ниманн-Пика Причина: Снижение активности фермента сфингомиелиназы. Тип наследования: Аутосомно-рецессивное. Клиника: Болезнь чаще проявляется в раннем возрасте. У ребенка увеличиваются лимфатические узлы, размеры живота, печени и селезенки; отмечаются рвота, отказ от пищи, мышечная слабость, снижение слуха и зрение. У 20-30% детей на сетчатке глаза обнаруживается пятно вишневого цвета (симптом "вишневой косточки"). Поражение нервной системы ведет к отставанию нервно-психического развития, глухоте, слепоте. Резко снижается устойчивость к инфекционным заболеваниям. Дети погибают в раннем возрасте. Диагностика: Выявление в плазме крови и спинномозговой жидкости повышенного содержания сфингомиелина. В периферической крови выявляются большие зернистые пенистые клетки Пика. Патогенез: В результате происходит накопление сфингомиелина в клетках печени, селезенке, мозге, ретикуло-эндотелиальной системе. Вследствие дегенерации нервных клеток нарушается деятельность нервной системы. Лечение: Симптоматическое. 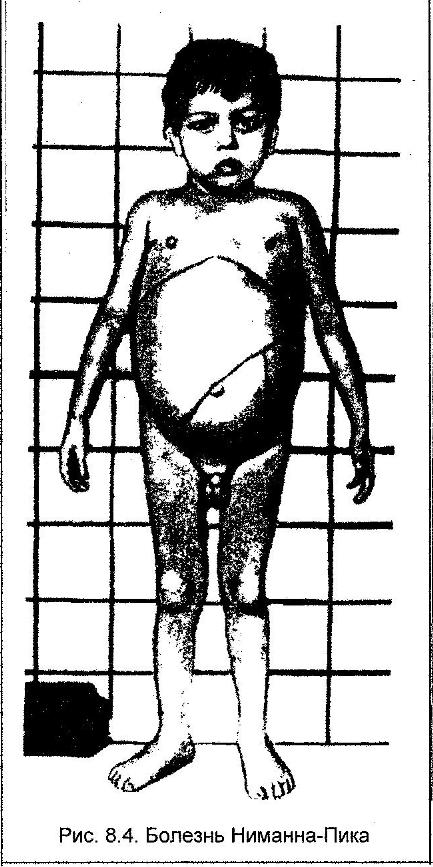 Болезнь Гоше Причина: Причиной болезни Гоше является дефект гена глюкоцереброзидазы. Глюкоцереброзидаза - это фермент, который помогает расщеплять глюкоцереброзид, находящийся в кровяных клетках-макрофагах. Клетки приобретают специфический вид так называемых "пенистых клеток", или клеток Гоше. Тип наследования: Аутосомно-рецессивное. Клиника: Клетки Гоше вначале накапливаются в печени, селезенке, костном мозге и иногда в легких. Когда это происходит, пораженные органы, особенно печень и селезенка, увеличиваются. Также при этом заболевании имеет место костная патология (атрофия, склероз, некроз). Клетки Гоше могут занимать значительный объем здорового костного мозга, часто приводят к спонтанным кровотечениям. Кроме того, наблюдаются такие изменения в крови, как анемия, тромбоцитопения (малое число тромбоцитов) и даже панцитопения (резкое снижение количества всех клеток в крови). Диагностика: Если подозревается болезнь Гоше, то диагноз может быть подтвержден анализом, который измеряет активность фермента глюкоцереброзидазы в крови. Такой анализ выполняется в нашей лаборатории медицинской генетики. Для этого нужна кровь с вены и через несколько дней будет получен результат. Патогенез: У больных поражены печень, селезенка. Происходят изменения в крови. Лечение: Лечение этой болезни возможно с помощью введения недостающего фермента.  Рахит, витамин D-зависимый Причина: В крови гипокальцемия, повышение содержания витамина D в 10-100 раз. Тип наследования: Х-сцепленный доминантный. Клиника: Патология обычно появляется в возрасте до 2 лет. Обращают на себя внимание следующие симптомы: замедление роста, моторного развития, мышечная гипотония, слабость. Могут быть патологические переломы, судороги и развитие тетании. Рентгенологически выявляются рахитоподобные изменения скелетной системы. Характерна гипофосфатемия, уровень кальция в крови в пределах нормы или ниже. Диагностика: Лабораторные исследования. Внешние признаки. Рентгенологические обследования. Патогенез: Изменение скелетной системы. Лечение: Диетотерапия. Альбинизм Причина: Врожденное отсутствие или инактивация энзима тирозиназы в эпителиальных клетках, в связи, с чем нарушается образование пигмента меланина. Тип наследования: Аутосомно-рецессивный. Клиника: Врожденное отсутствие пигментации кожи, волос, радужной оболочки, в результате нарушения обмена фенилаланина и тирозина. Светобоязнь, красный зрачковый рефлекс, высокая чувствительность к солнечным лучам. В связи с отсутствием пигмента в фоторецепторах сетчатки отмечается избыточный распад зрительного пигмента родопсина, поэтому больные плохо видят днем. Отсутствует бинокулярное зрение, кожа розовато-красная. Диагностика: Основана на клинических данных. Профилактика: Если оба родителя гомозиготны по одному и тому же признаку (оба альбиносы), то в их браке рождаются исключительно пораженные дети. Лечение: Не существует, полного выздоровления не бывает.  Синдром Марфана (арахнодактилия) Синдром Марфана (арахнодактилия)Причина: Изменение обмена мукополисахаридов приводит к нарушению образования коллагена. Тип наследования: Аутосомно-доминантный. Клиника: Высокий рост, длинные тонкие конечности, длинные пальцы рук и ног (паучьи пальцы) и относительно укороченное туловище, килевидная или воронковидная грудная клетка, лицо имеет птичье выражение, тонкий нос. Изменение строения глаз. Слабость связочного аппарата, поэтому наблюдаются вывихи в суставах. Интеллект в норме. Заболевание постепенно прогрессирует, симптомы становятся более отчетливыми. Патогенез: Аномалии сердечно-сосудистой системы (пороки сердца, аневризма аорты, недостаточность митрального клапана). Поражения желудочно-кишечного тракта и мочевыделительной системы. Диагностика: Клиническое обследование и биохимический анализ мочи на наличие повышенного количества оксипролина - аминокислоты, входящей в состав коллагена. Лечение: Симптоматическое. Оперативное лечение вывиха хрусталика, торакопластика при деформации грудной клетки. Курс массажа и лечебной физкультуры. 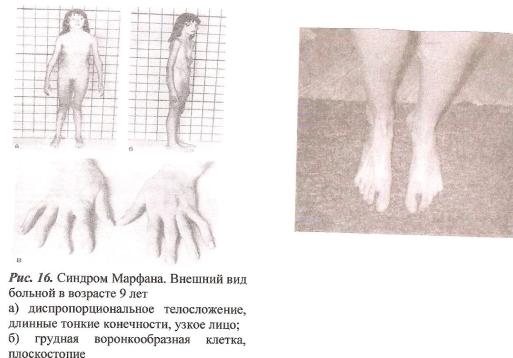 Мукополисахаридоз Причина: Мукополисахаридозы представлены целой группой наследственных заболеваний соединительной ткани. Для них характерно нарушение в организме метаболизма кислых гликозоаминогликанов, что связано с недостаточностью лизосомальных ферментов. Тип наследования: Аутосомно-рецессивный. Клиника: Признаками болезни служат: замедление роста, короткая шея и туловище, деформация костей, снижение интеллекта, грубые черты лица с крупными губами и языком, пупочные и паховые грыжи, пороки сердца, нарушение психического развития с отставанием от нормы. Диагностика: Лабораторные исследования, внешние признаки. Патогенез: Патологические продукты обмена откладываются в соединительной ткани, в печени, селезенке, роговице и в клетках центральной нервной системы. При мукополисахаридозах поражаются опорно-двигательный аппарат, внутренние органы, глаза, нервная система. Лечение: диетотерапия, физиопроцедуры (электрофорез, магнитотерапия, массаж, лечебная физкультура и др.), гормональные и сердечнососудистые средства.  Серповидно-клеточная анемия Причина: Генные мутации полипептидной цепочки гемоглобина; в результате наблюдается преждевременный гемолиз и распад эритроцитов, обусловленный низкой способностью гемоглобина связывать и переносить кислород. Тип наследования: Аутосомно-рецессивный, кодоминантный. Клиника: Заболевание носит семейный характер и проявляется в тяжелой и легкой форме. Тяжелая форма обусловлена гомозиготным рецессивным состоянием аномального гена. Больные погибают в раннем детстве или при достижении половой зрелости от тяжелой гемолитической анемии с низким гемоглобином. Легкая форма - при гетерозиготном носительстве того же гена. У лиц гетерозиготных по гену гемоглобина в эритроцитах содержится как аномальный, так и нормальный гемоглобин, заболевание у них проявляется в очень легкой форме лишь в условиях кислородной недостаточности. Характерна бледность кожи и слизистых оболочек, желтушность, усиливающаяся с возрастом. Патогенез: Измененная последовательность аминокислотных остатков в гемоглобине приводит к замедлению электрофоретической подвижности; повышенная вязкость крови, гемолиз. Приступы болей в суставах, скелете, мышцах. Расстройство кровообращения в мозге, почках и селезенке вследствие образования тромбов. Диагностика: Клиническое обследование и биохимический анализ крови (эритроциты больного имеют вытянутую серповидную форму). Лечение: Требуется больным с острым периодом болезни. Для снятия болевого синдрома используются анальгетики. Для разведения эритроцитов больному переливают эритроцитарную плазму здорового человека. 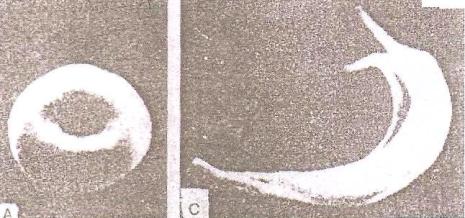  Электронная микрофотография эритроцитов: А)нормальная форма; Б, В- больные эритроциты L-талассемия Причина: Мутация регуляторных глобиновых генов, вследствие чего наблюдается нестабильный синтез и дисфункция матричной РНК. Дефицит продукции L-глобиновых цепей приводит к снижению содержания нормального гемоглобина. От количества отсутствующих генов зависит тяжесть заболевания. Тип наследования: Аутосомно-доминантный, кодоминантный. Клиника: При гомозиготности полное или частичное отсутствие гемоглобина и наличие гемоглобина, не способного переносить кислород. Плод гибнет внутриутробно. При гетерозиготной форме - гемоглобинопатия, хроническая гемолитическая анемия средней тяжести. Патогенез: При гетерозиготной форме - морфологические изменения эритроцитов, вследствие чего развивается кислородная недостаточность в тканях и органах организма. Диагностика: Клиническое обследование и биохимический анализ крови, при котором выявляются морфологические изменения эритроцитов. Лечение: Препараты фолиевой кислоты и витамины группы В. Предупреждение браков между гетерозиготными носителями генов талассемии и рекомендации отказа от потомства. Нейрофиброматоз (болезнь Реклингхаузена)Причина: У больных в печени, почках и слизистой кишечника накапливается большое количество гликогена. Превращение его в глюкозу не происходит, т.к. отсутствует фермент глюко-6-фосфатаза, регулирующий уровень глюкозы в крови. В результате у больного развивается гипогликемия, в печени, почках и слизистой кишечника накапливается гликоген. Тип наследования: Аутосомно-доминантный. Клиника: У больных на коже появляются мелкие опухоли (ней-рофибромы) - от единичных до нескольких сотен. Они могут локализоваться повсюду, в том числе и на слизистых оболочках ротовой полости и языка. Описаны пациенты с числом нейрофибром до 10000 и более. Нейрофибромы представляют собой мягкие узелки, при надавливании как бы проваливающиеся в кожу — симптом «кнопки звонка». Подкожные узелки располагаются по ходу нервных стволов (округлые бусинки диаметром 1 — 2 см, подвижные, не прикрепленные к коже). Помимо этого, у некоторых больных развиваются диффузные массивные опухолевидные образования. Веснушки в подмышечных и паховых складках, пятнистая гиперпигментация кожи верхней части груди и промежности относятся также к частым симптомам этого заболевания. Затруднения в обучении наблюдаются у 30 % больных. Умственная отсталость не глубокая и не прогрессирующая. Диагностика: По клинической картине. Лабораторные исследования. Патогенез: Почти у всех больных наблюдаются изменения костной системы — кифоз, сколиоз, псевдоартрозы, локальный гигантизм, неспецифические черепно-лицевые аномалии. Лечение: Симптоматическое. 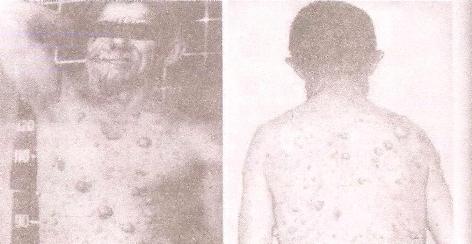 Наследственный микросфероцитоз — гемолитическая анемия Минковского-Шоффара. Причина: Заболевание обусловлено генетическими аномалиями эритроцитов и связано с врожденной недостаточностью липидов их оболочки. В результате повышения проницаемости мембраны в клетку проникают ионы натрия и теряется АТФ. Эритроциты принимают сферическую форму. Измененные эритроциты разрушаются в селезенке с образованием токсического белка — билирубина. Болезнь может протекать в двух формах — хронической и острой, при которой усиливается гемолиз. Клиника: Желтуха, анемия, спленомегалия (разрыв селезенки), изменение скелета. У детей в первые месяцы жизни часто возникает "ядерная желтуха". Причина — поражение ядер головного мозга за счет высокого содержания билирубина. Для больных характерно увеличение селезенки и печени, деформация скелета; башенный череп, готическое небо, нарушенное расположение зубов. Диагностика: Исследуется кровь. Характерными признаками являются обнаружение микросфероцитов, снижение осмотической стойкости эритроцитов и др. Патогенез: В старшем возрасте высокий уровень билирубина приводит к образованию камней и развитию желчекаменной болезни. Лечение: Симптоматическое. Синдром Беквита - Видемана Причина: Отмечается гипогликемия, сочетающаяся с рядом соматических изменений, что может являться причиной умственной отсталости, макроглоссия и омфалоцеле (пуповинная грыжа или расхождение прямых мышц живота). Язык крупный, иногда не помещается в ротовой полости, вследствие чего рот открыт. У новорожденного могут быть затруднены сосание и дыхание. Тип наследования: Аутосомно-доминантный. Клиника: гигантизм (макросомия) с увеличением мышечной массы и подкожно жирового слоя отмечается либо с рождения, либо развивается постнатально. Череп с выступающим затылком, нарушение прикуса. Диагностика: По внешним признакам. Патогенез: Внутренние органы увеличены. Пуповинные грыжи. Лечение: Симптоматическое. При грыжах- хирургическое. Синдром Вильямса, «лицо Эльфа». Причина: У детей повышенный уровень кальция в сыворотке крови. Тип наследования: Аутосомно-доминантный. Клиника: У детей отмечается своеобразие черт лица, что создается пухлыми опущенными вниз щеками, большим ртом с полными губами, маленьким подбородком, широким и сдавленным в висках лбом, своеобразным разрезом глаз с припухлостью вокруг орбит, характерной звездчатостью радужки, коротким носом с открытыми вперед ноздрями и закругленным тупым концом. Часто наблюдаются голубоватые склеры и ярко-голубые радужки. Типичны эпикант, открытый рот и оттопыренные уши. Ни один из признаков не является постоянным, а их сочетание создает сходство между больными. Характерным признаком считают также редкие зубы, пороки сердца и умственная отсталость. В раннем возрасте дети отличаются выраженной соматической ослабленностью, отстают в росте и массе тела. В дальнейшем у многих из них может развиться тучность. Обращают на себя внимание длинная шея, узкая грудная клетка, низкая талия. Х-образные ноги. Наблюдаются плоскостопие, иногда косолапость и повышенная разгибаемость суставов. Диагностика: Лабораторные исследования на уровень кальция в крови; по внешним признакам. Патогенез: Мышечная гипотония. Пороки сердца, нарушения опорно-двигательного аппарата. Умственная отсталость. Лечение: С раннего возраста дети нуждаются в общеоздоровительных и лечебно-коррекционных мероприятиях, с ограничением потребления кальция. 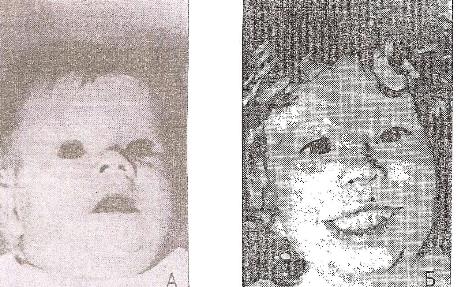 Гликогеноз ( 1 тип - Болезнь Гирке) Причина: У больных в печени, почках и слизистой кишечника накапливается большое количество гликогена. Превращение его в глюкозу не происходит, т.к. отсутствует фермент глюко-6-фосфатаза, регулирующий уровень глюкозы в крови. В результате у больного развивается гипогликемия, в печени, почках и слизистой кишечника накапливается гликоген. Тип наследования: Аутосомно-рецессивный. Клиника: Сразу после рождения главными, симптомами болезни являются гликогемические судороги и гепатомегалия (увеличение печени). С 1-го года жизни отмечается задержка роста. Характерный вид больного: большая голова, "кукольное лицо", короткая шея, выступающий живот. Кроме того, отмечаются носовые кровотечения, задержка физического и полового развития, мышечная гипотония. Интеллект при этом нормальный. Диагностика: Лабораторные исследования на уровень гликогена в крови, внешние признаки. Патогенез: В крови повышается уровень мочевой кислоты, так что с возрастом может развиться подагра. Лечение: Используется диетотерапия: частый прием пищи, повышенное содержание углеводов и ограничение жиров в диете.  Гликогеноз (II тип — болезнь Помпе) Причина: Гликоген накапливается как в печени, так и в скелетных мышцах, миокарде, легких, селезенке, надпочечниках, стенках сосудов, в нейронах. Тип наследования: Аутосомно-рецессивный. Клиника: У новорожденных спустя 1-2 месяца появляется мышечная слабость, дефицит 1,4-глюкозидазы в печени и в мышцах. В этот же период возникают увеличение сердца и патологическое увеличение языка. Нередко у больных развивается тяжелая форма пневмонии из-за накопления секрета в дыхательных путях. Дети погибают на первом году жизни. Диагностика: Возможна еще до рождения ребенка. С этой целью определяют активность фермента 1,4-глюкозидазы в амниотической жидкости и ее клетках. Патогенез: Происходят патологические изменения внутренних органов. Лечение: Не существует. Амавротическая идиотия (болезнь Тея-Сакса) Причина: Болезнь связана с нарушением липидного обмена. Для нее характерно отложение в клетках мозга, печени, селезенки и других органах липида ганглиозида. Причина - снижение активности фермента гексозаминидазы А в организме. Тип наследования: Аутосомно-рецессивный. Клиника: Болезнь проявляется в первые месяцы жизни. Ребенок становится вялым, малоподвижным, безразличным к окружающим. Задержка психического развития приводит к снижению интеллекта до степени идиотии. Отмечается мышечная гипотония, судороги, характерный симптом "вишневой косточки" на сетчатке глаза. К концу первого года жизни наступает слепота. Позднее развивается полная обездвиженность. Смерть наступает в 3-4 года. Диагностика: По внешним признакам. Лабораторная диагностика. Патогенез: Происходит разрушение аксонов нервных клеток, атрофия зрительных нервов. Лечение: Не существует.  Синдром Фабри Причина: Синдром Фабри (диффузная ангиокератома туловища) относится к группе липоидозов. Синдром обусловлен недостаточностью фермента альфа-галактозидаза А при мутационном поражении гена. Тип наследования: наследуется по рецессивному, сцепленному с полом типу. Клиника: заболевание проявляется поражениями кожи, сосудистой системы глаз и внутренних органов. Развитие сосудистых поражений кератизинированных узелков на коже губ, щек, подкрыльцовой впадины, концевых фаланг пальцев, живота, ягодиц, груди и других частей туловища в виде мелких сосудистых пятен с окраской различной интенсивности, возвышающихся над кожей. При травмировании узелки кровоточат. Диагностика: По внешним признакам. Лабораторная диагностика. Патогенез: повышение артериального давления, изменения сосудов сетчатки, помутнение роговой оболочки глаз, почечная недостаточность. Отмечается увеличение фосфатов и липидов в крови. Синдром Робертса, тетрафокомелия с расщелиной губы и неба. Причина: Генные изменения. Тип наследования: Аутосомно-рецессивный. Клиника: Для тяжелой формы синдрома типичны тетрафокомелия, двусторонняя расщелина губы и неба, гипертелоризм, микроцефалия. Поражение конечностей носит обычно симметричный характер, при этом верхние конечности поражены, как правило, в большей степени, чем нижние. Тяжесть поражения варьирует от фокомелии до различной степени редукции конечностей с деформацией и гипоплазией различных участков. Чаще всего отсутствуют кости предплечья и голени, примерно в половине всех случаев отсутствуют проксимальные отделы конечностей (плечевые и бедренные кости). Длина конечностей зависит от степени гипоплазии. Обычно у пораженных длина верхних конечностей составляет половину их нормальной длины, а длина нижних конечностей колеблется от половины нормальных размеров до почти нормальной длины. Диагностика: Внешние признаки. Патогенез: Изменения скелетной системы. Лечение: Не существует. 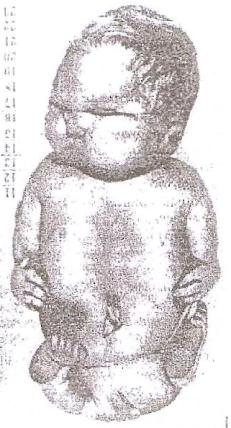 Синдром умственной отсталости с ломкой х-хромосомы (синдром Мартина-Белл) Причина: Мутантный ген картирован. Он расположен в Xq28. У больных в этом участке Х-хромосомы обнаружено значительное увеличение (до 1000 и более) нестабильных тринуклеотидных повторов (CGG). В этом участке происходит снижение конденсации хроматина при культивировании лимфоцитов на специальных питательных средах, лишенных фолиевой кислоты. Тип наследования: Рецессивный, сцепленный с Х-хромосомой. Клиника: Ряд внешних особенностей больных неспецифичен. Но определенное диагностическое значение имеют следующие признаки: высокий рост; крупные кисти и стопы; высокий выступающий лоб. Удлиненное лицо с уплощенной срединной частью; толстые губы, нижняя часть вывернута; клювовидный нос; высокое арковидное нёбо; «оттопыренные», увеличенные в размерах уши. Гиперэластичность кожных покровов; переразгибаемость суставов, плоскостопие. Патогенез: С наступлением пубертатного периода размер тестикул существенно увеличивается. Увеличение размеров тестикул происходит за счет избыточного роста соединительной ткани и накопления в них жидкости. Диагностика: В настоящее время возможна пренатальная диагностика этого заболевания. Лечение: Этиологической терапии пока не существует. 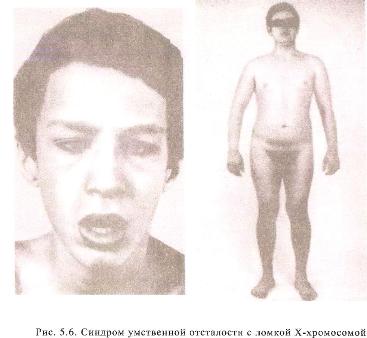 Псевдогипертрофическая мышечная дистрофия Дюшенна Причина: Заболевание обусловлено нарушением синтеза белка дистрофина. Ген дистрофина локализован в коротком плече Х-хромосомы, клонирован и секвенирован. Тип наследования: Х- сцепленный рецессивный. Клиника: Основная симптоматика заболевания заключается в прогрессирующем нарастании мышечных дистрофических изменений с постепенным обездвиживанием больного. У детей до трехлетнего возраста диагностировать заболевание достаточно сложно. Известно, что эти дети несколько отстают в моторном развитии на первом году жизни — позже начинают сидеть, ходить. Никогда не бегают и не прыгают. Классическая картина заболевания проявляется у детей 3-5 лет. Одним из первых признаков является уплотнение икроножных мышц и постепенное увеличение их объема за счет разрастания соединительной и жировой ткани. В терминальной стадии слабость мышц может распространяться на мышцы лица, шеи, глотки. В развитой стадии болезни имеются такие характерные симптомы, как «утиная» походка, выраженный поясничный лордоз (см. рис.), крыловидные лопатки. Типичны ранние мышечные контрактуры. Псевдогипертрофии могут развиваться также и в ягодичных и дельтовидных мышцах, мышцах языка и живота. Очень часто страдает сердечная мышца. Выявляются нарушения сердечного ритма, расширение границ сердца, изменения ЭКГ. Острая сердечная недостаточность — наиболее частая причина смерти. Примерно у 50 % детей отмечается снижение интеллекта — от пограничных состоянии до выраженной дебильности. Погибают больные, как правило, на третьем десятилетии жизни, а к 14 —15 годам они, обычно, обездвижены. Диагностика: Клинические исследования. Эхография. Патогенез: Мышечные дистрофии характеризуются дегенеративными изменениями в поперечно-полосатой мускулатуре без первичной патологии периферического мотонейрона. Развивается сердечная недостаточность. Лечение: Симптоматическое.   Мышечная дистрофия Дюшенна.(дистрофия икроножных мышц) Синдром Прадера-Вилли Причина: инактивация генов области g11-13 хромосомы 15 отцовского происхождения. Тип наследования: Аутосомно-доминантный. Клиника: выделяют 2 группы признаков - те, которые наблюдают до трех летнего возраста, и появляющиеся в старшем возрасте. Первые признаки возникают с рождения. Главные среди них – мышечная гипотония, трудности вскармливания и малый вес при рождении. Иногда до 6месячного возраста у ребенка наблюдается повышенная сонливость или даже приступы летаргии. Для всех больных характерна дисморфия – широкий бифронтальный диаметр, миндалевидный разрез глаз, опущенные углы рта. После 6 месяцев может возникнуть гиперфагия. Усиление аппетита приводит к быстрой прибавке в весе и ожирению. Избыточный вес и нарушение обмена веществ часто провоцируют развитие сахарного диабета. Патогенез: гипопигментация кожи, часто обнаруживается снижение чувствительности к боли; харектерен гипогонадизм- гипоплазия полового члена у мужчин и малых половых губ у женщин. Диагностика: анализ крови, ДНК-анализ. Лечение: симптоматическое. 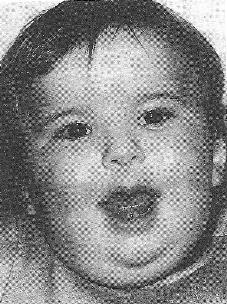 Синдром Энгельмана Причина: инактивация генов области g11-13 хромосомы 15 материнского происхождения. Тип наследования: Аутосомно-доминантный. Клиника: Болезнь проявляется грубой задержкой психомоторного развития, выраженной олигофренией и недоразвитием речи. Больные поздно начинают ходить и имеют своеобразную походку (на широко расставленных ногах с согнутыми в локтевых суставах руками), напоминающую движения механической куклы. Типичным проявлением заболевания являются приступы насильственного немотивированного смеха. Сочетание этих двух симптомов привело к тому, что Синдром Энгельмана часто называют «синдромом лица счастливой куклы». Патогенез: судороги, выраженные расстройства координации движений, мышечная гипотония, косоглазие. В некоторых случаях имеет место гипопигментация волос и кожи. Диагностика: По внешним признакам.(молекулярно-генетические и цитогенетические методы) 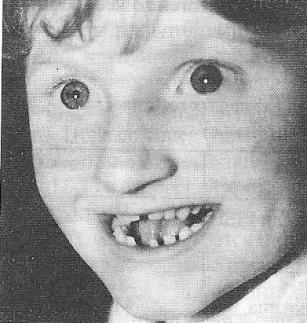 Ахондроплазия Причина: Из-за сниженной способности больных иметь потомство в 80-95% случаев это заболевание связано с заново возникающими мутациями. Как большинство доминантных мутаций, ахондроплазия встречается редко. Тип наследования: Аутосомно-доминантный. Клиника: низкий рост (при рождении 46-48 см., у взрослого 120-130см.), большой череп с выступающим затылком, запавшая переносица. Конечности укорочены, кисти широкие и короткие, пальцы расположены в виде тризубца, лордоз. Это одна их наследственных болезней костной системы, клиническая картина её обусловлена аномальным ростом и развитием хрящевой ткани, главным образом в эпифизах трубчатых костей и оснований черепа. Хрящевые зоны этих костей могут быть гипопластичными или аномально гиперплазированными, результатом чего является резкое недоразвитие костей в длину. О биохимической природе этой формы хондродистрофий ничего неизвестно, если не считать сведений о различных отклонениях активности ряда ферментов, значение которых остаётся пока неясным. Патогенез: Патология роста костей определяет характерную клиническую картину, полностью оформляющуюся в половозрелом возрасте:
Диагностика: По внешним признакам. Лечение: Не существует. 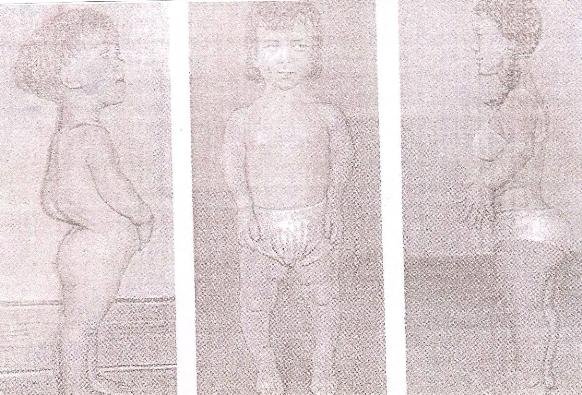 Полидактилия Причина: Генная мутация. Тип наследования: Аутосомно-доминантный. Клиника: Увеличение количества (до 8-12) пальцев на кистях и стопах. В случае преаксиальной полидактилии дополнительный палец находится со стороны 1-го пальца с дупликацией всех или только части его составных элементов. При полидактилии 2-го пальца происходит его удвоение. Дополнительный палец расположен со стороны локтевой кости на руке и со стороны малоберцовой кости на ноге. Часто входит в состав множественных врожденных пороков развития. Патогенез: Специфичное изменение внутренних органов не характерно. Диагностика: Клиническое обследование. Лечение: Хирургическое устранение дефекта. Адреногенитальный синдром (врожденная гиперплазия коры надпочечников) Причина: Адреногенитальный синдром (АТС) относится к группе наследственных нарушений биосинтеза стероидных гормонов. Тип наследования: Аутосомно-рецессивный. Клиника: В клинике практически с момента рождения наблюдается обильное срыгивание, рвота, сонливость, нарушение периферического кровообращения, потеря массы тела. Биохимическое исследование выявляет гиперкалиемию, гипонатриемию, ацидоз. Диагноз в периоде новорожденного и в раннем детстве можно заподозрить только у девочек, в связи с неправильным строением наружных половых органов. Симптомы вирилизации наружных гениталий колеблются от незначительной гиперплазии клитора до правильно сформированного пениса, с открытием мочеиспускательного канала на головке, с наличием мошонки, но с отсутствием в ней тестикул. Внутренние половые органы сформированы правильно и соответствуют биологическому полу (кариотип 46, XX). Они представлены маткой и яичниками. Правильное определение пола ребенка в первые дни жизни позволяет в дальнейшем избежать тягостных формальностей, связанных со сменой пола. У мальчиков этот диагноз можно предположить лишь в 3-6 летнем возрасте, когда начинается преждевременное половое развитие — гиперпигментируется мошонка, появляется половое оволосение, грубеет голос. На первом десятилетии жизни дети, больные АТС, значительно обгоняют своих сверстников в росте, затем темпы роста снижаются. Рост взрослых людей с этим заболеванием составляет около 140 см без соответствующего лечения. Поздняя форма проявляется в пубертатном периоде. У девочек возникает незначительная гиперплазия клитора, гирсутизм, ускорение костного возраста, различные нарушения менструального цикла. Симптомами заболевания у мальчиков могут быть лишь преждевременное оволосение наружных гениталий и снижение темпов роста за счет раннего закрытия зон роста. Латентная форма практически не имеет клинических проявлений, но в сыворотке крови отмечается умеренное повышение предшественников кортизола. Диагностика: Биохимические исследования крови. Патогенез: Неправильное формирование половых органов. Лечение: Симптоматическое. Гормонотерапия. 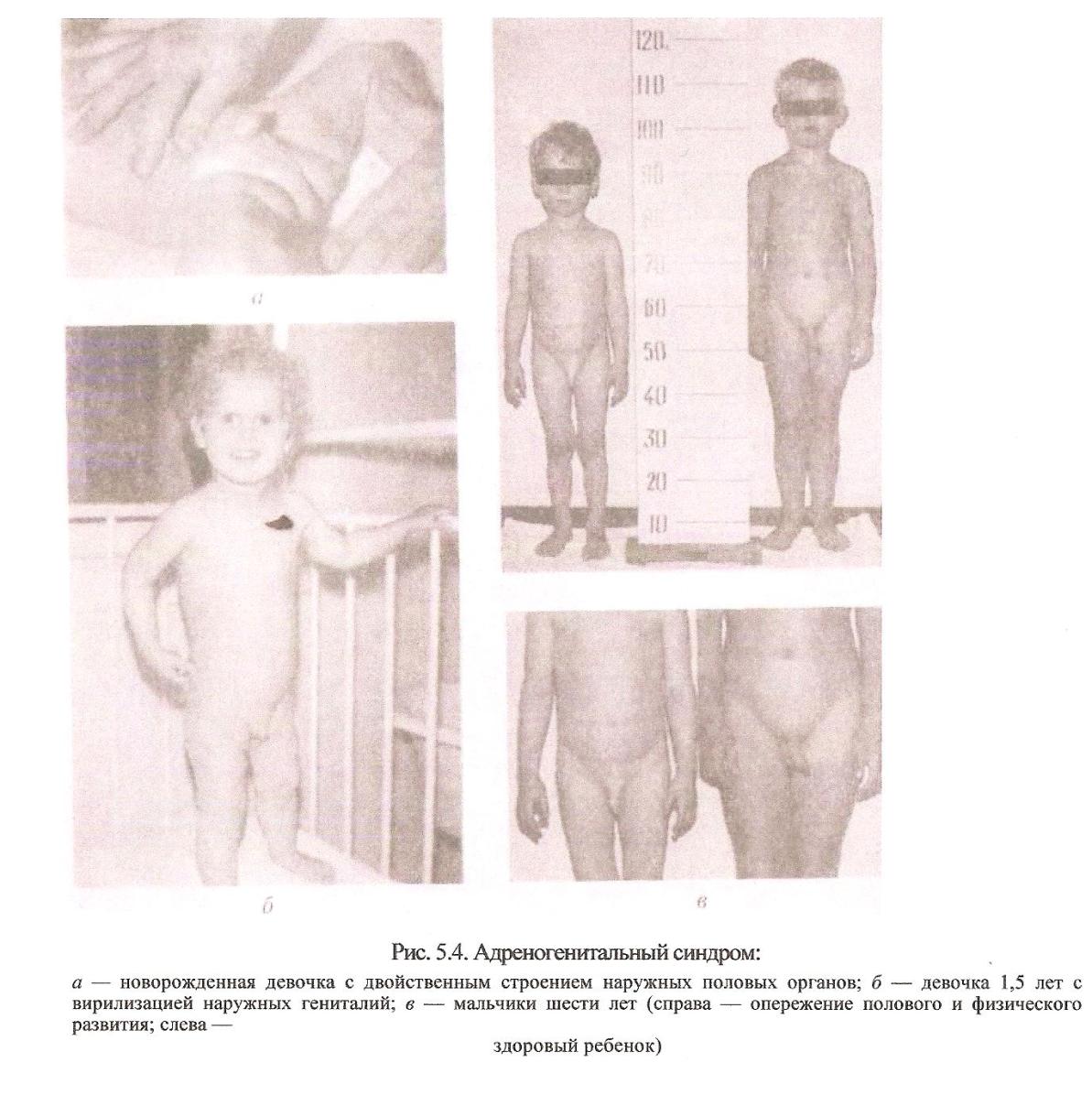 Синдром Холт-Орама Синдром Холт-ОрамаПричина: Множественные врожденные пороки развития. Тип наследования: Аутосомно-доминантный. Клиника: Клиническая картина характеризуется аномалиями верхних конечностей и врожденными пороками сердца. Часто наблюдаются и другие скелетные изменения: гипоплазия лопаток и ключиц, сколиоз, воронкообразная деформация грудины и др. Диагностика: Диагноз ставится на основании клинико-генеалогических данных. Также проводятся рентгенография и эхография. Патогенез: Дефекты межпредсердной и межжелудочковой перегородок, открытого аортного протока, стеноза легочной артерии, пролапса митрального клапана и другие. Лечение: Симптоматическое, при необходимости хирургическое.  Муковисцидоз Причина: исходное наследственное поражение экзокринных желез и железистых клеток организма: секретирующих клеток бронхов, поджелудочной железы, кишечника, потовых желез и печени. Тип наследования: Аутосомно-рецессивный. Клиника: Различаются несколько форм муковисцидоза. У новорождённых может развиться непроходимость кишечника, обозначаемая как мекониальный плеус. Это проявление тяжёлой формы муковисцидоза, встречающееся у 10-20% больных муковисцидозом новорождённых. Патологические изменения в лёгких при муковисцидозе имеют место у 85-95% всех больных и проявляются либо в преимущественно легочной форме этой болезни, либо в смешанной лёгочно-кишечной форме. Заболевание начинается в грудном возрасте, как острое воспалительное. В начале развивается острая пневмония. В последующем пневмонии становятся повторными, затяжными и хроническими. Обязательным компонентом патологии становятся бронхиты, позднее развивается энфизема и пневмосклероз. При кишечной форме муковисцидоза доминируют симтомы нарушения пищеварительных функций поджелудочной железы и кишечника. Нарушенная усвояемость пищи приводит к дистрофии. У 20% больных муковисцидозом в клинической картине могут преобладать симптомы гепатита, жировой дистрофии, а позже и билиарного цирроза печени. Патогенез: Общим моментом патологии желез внешней секреции является нарушение их функций - выделение густого, изменённого по составу секрета. Это ведёт к застойным изменениям в соответствующих органах, последующим воспалительным и склеротическим процессам. В секретах организма при муковисцидозе изменяется соотношение фракций его белково-углеводных компонентов; наблюдается снижение реабсорбции электролитов в выводных протоках потовых желез; поджелудочная железа не выделяет нужного количества фермента. Диагностика: Клинические и лабораторные исследования. Лечение: Медикаментозное, хирургическое - в зависимости от симптомов. Хромосомные болезни Хромосомные болезни, или синдромы - это группа врожденных патологических состояний, проявляющихся множественными пороками развития, различающихся по своей клинической картине, часто сопровождающихся тяжелыми нарушениями психического и соматического развития. Основной дефект - различные степени интеллектуальной недостаточности, что может осложняться нарушениями зрения, слуха, опорно-двигательного аппарата, более выраженными, чем интеллектуальный дефект, расстройствами речи, эмоциональной сферы и поведения. Диагностические признаки хромосомных синдромов можно разделить на три группы:
Хромосомные заболевания не подчиняются менделеевским закономерностям передачи заболевания потомству и в большинстве случаев обнаруживаются спорадически, являясь следствием мутации в половой клетке одного из родителей. Хромосомные болезни могут быть унаследованы, если мутация имеется во всех клетках родительского организма. 1 2 |