хим сессия 1-29. Наука древних первый аналитический прибор весы
 Скачать 3.43 Mb. Скачать 3.43 Mb.
|

|
История становления аналитической химии. Впервые было введено в 1661 г. английским химиком Робертом Бойлем. Периоды развития аналитической химии делим на 6 групп: Наука древних первый аналитический прибор — весы — известен с глубокой древности у римского историка Плиния описана методика анализа золота, еще раньше об оценке содержания золота писал император Вавилона в древности умели определять концентрацию по удельному весу; само понятие “удельный вес” известно по крайней мере со времен Архимеда. По-видимому, вторым по времени появления аналитическим прибором был ареометр, он описан в трудах древнегреческих ученых. в произведении Теофраста "О камнях" говорится об определении золота с помощью так называемого пробного, или пробирного, камня Алхимия (IV—XVI вв.) отработан метод определения золота и серебра, основанный на «пробирной плавке» — плавлении в присутствии восстановителя и металла-носителя (обычно свинца), в расплаве которого хорошо растворяются драгоценные металлы получил дальнейшее развитие метод пробного камня, в средние века стали использовать шкалу из 24 игл с разным содержанием золота для анализа стали использовать растворы Иатрохимия (XVI—XVII вв.) – наука о приготовление лекарств и налаживания некоторых химических производств (целительная, лекарственная алхимия) Роберт Бойль ввел термин «химический анализ», использовал экстракты растений (лакмус, фиалка и др.) и животных тканей для определения кислотности и щелочности растворов появились новые способы обнаружения веществ, основанные на переводе их в раствор, были введены понятия «осаждение», «осадок» Эпоха флогистона (XVII—XVIII вв.) Термин «ФЛОГИСТОН» введён в 1667 году Иоганном Бехером и в 1703 году Георгом Шталем для объяснения процессов горения создателями газового анализа были Г. Кавевдиш, Дж. Пристли, К. Шееле с их именами связано открытие кислорода и водорода К. Шееле получил щавелевую кислоту, которую предложил впервые как реагент на кальций А. Маргграф, начал использовать микроскоп в химическом анализе, ввел новые методы, в том числе способ определения серебра с помощью хлорида шведский химик Т. Бергман впервые провел различие между качественным и количественным анализом, обобщил накопленный к тому времени материал о применении паяльной трубки в анализе, установил влияние углерода и фосфора на свойства железа Период научной химии (XIX—XX вв.) А. Л. Лавуазье – открытие кислородной теории горения, закона сохранения вещества, различия между элементами и соединениями И. В. Рихтер - становление законов стехиометрии Дальтон - закон кратных отношений, шкала атомных весов Я. Берцелиус - определил атомные веса почти всех известных тогда элементов, ввел символы элементов, химические формулы, активно проводил аналитические на основе правил стехиометрии. Берцелиус стоял у истоков метрологии анализа. Он оценивал ошибки определений, разработал точные методы взвешивания, ему принадлежит методика определения платиновых металлов, пытался создать новую схему качественного анализа. При анализе силикатов Берцелиус использовал возгонку хлоридов для разделения металлов к середине XIX в. оформились титриметрические, гравиметрические методы, способы элементного органического анализа, методы газового анализа Современный период 1903г М.С. Цвет - открытие хроматографии и последующее создание разных вариантов хроматографического метода А. Мартин и Р. Синдж – метод распределительной хроматографии (Нобелевская премия по химии) А. Тизелиус - исследования по электрофорезу и «адсорбционному анализу» метод полярографии, метод комплексонометрического титрования появилось много физических и химических методов анализа — масс- спектрометрические, рентгеновские, ядерно-физические, новые варианты электрохимических методов, интенсивно развивались фотометрические методы А. Уолш, К. Алкемаде, Б. В. Львов, 50-е годы - атомно-абсорбционный метод (Нобелевская премия) расширяется арсенал методов анализа, автоматизация и математизация анализа; создание приемов и средств локального, неразрушающего, дистанционного, непрерывного анализа; подход к решению задач о формах существования компонентов в анализируемых пробах; появление новых возможностей для повышения чувствительности, точности и экспрессности анализа; расширение круга анализируемых объектов, использование компьютеров, лазеров, лабораторных роботов; поднялась роль аналитического контроля объектов окружающей среды 2. Виды химического анализа и его стадии Качественный анализ – обнаружение и идентификация компонентов Количественный состав – определение абсолютных количеств обнаруженных компонентов Стадии химического анализа Отбор пробы для анализа Подготовка пробы к анализу Оценка качества Измерение интенсивности аналитического сигнала Обработка результатов. 3. Системы анализа Сульфидная классификация основана на различной растворимости в воде сульфидов, хлоридов, карбонатов и гидроксидов. В 1871г. Н.А.Меншуткиным была разработана сероводородная (сульфидная) система анализа. Недостатки: - полный анализ требует много времени (25-30ч) - ядовитость h2s Аммиачно-фосфатная аналитическая классификация катионов В 1971г. на кафедре аналитической химии МХТИ им.Д.И.Менделеева разработана и апробирована аммиачно – фосфатная система анализа. В основу положена различная растворимость фосфатов в воде, водных растворах кислот, щелочей и аммиака. Кислотно-основный метод анализа катионов(Бесков и Слизковская) основан на различной растворимости в воде хлоридов, сульфатов и гидроксидов, а также растворимости последних в избытке раствора гидроксида натрия или в водном растворе аммиака. Соляная и серная кислоты, раствор NаОН и водный раствор аммиака являются групповыми реагентами. Достоинства классификации: используются основные свойства катионов, группы катионов практически полностью соответствуют группам периодической системы элементов Д.И. Менделеева, быстрота выполнения анализов, широкое применение систематического и дробного хода анализа. Недостатки: включает не все известные элементы, недостаточно отражены свойства гидроксидов катионов 4 и 5 групп, условия их осаждения. 4. ДРОБНЫЙ АНАЛИЗ Дробный анализ – это обнаружение определенных компонентов с помощью селективных и специфических реакций в отдельных порциях анализируемого раствора, проводимое в произвольной последовательности. 5. Методы анализа Все средства и методы а.х. делятся на три большие группы: 1. химические методы – в основе определенные химические реакции; 2. физико-химические методы – регистрация физико химических свойств веществ, например, электрохимический (измерение силы тока, потенциала), оптические (изучение специфики поглощения или излучения света), люминесценция и д.р.; 3. физические методы – основаны на измерении физических характеристик веществ: рентгеноструктурный анализ, радиоактивационный анализ Можно предложить и другие классификации видов анализа – например: изотопный, вещественный, молекулярный, структурно-групповой, фазовый – основанные на природе обнаруживаемых или определяемых частиц. 6   . Классификация методов: по цели анализа; по аналитическому сигналу; по объектам анализа; по массе или объему объекта, взятого для анализа  7. Закон действия масс. Константы равновесия обратимой химической реакции Зависимость скорости от концентрации реагирующих веществ выражается законом действия масс: скорость химической реакции прямо пропорциональна концентрациям реагирующих веществ (1867г. К.М.Гульдбергом и П.Вааге). Например, для обратимой химической реакции аА + вВ ↔ сС + dD (1) скорость можно выразить как произведение равновесных концентраций: ν1 = k1 [A]a[B]в, (2) где k1 – константа скорости прямой реакции. К  аждая аналитическая реакция является обратимой, только иногда эта обратимость проявляется в такой ничтожной степени, что ею можно пренебречь. Обратимые реакции недоходят до конца, но приводят к установлению хим.равновесия, при котором в растворе присутствуют как исходные, так и образовавшиеся в результате реакции вещества. В момент установления равновесия скорости прямой и обратной реакций сравниваются, т. е. если ν2 – скорость обратной реакции: 8  . Термодинамическая, реальная и условная константа равновесия Т 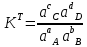 ермодинамические константы равновесия (табличные) можно использовать для вычисления рН раствора или растворимости осадка. Но они пригодны для расчётов равновесий только в идеальных системах, т. е. при условии, что поведение исходных веществ и продуктов реакции не осложняется электростатическими взаимодействиями или конкурирующими химическими реакциями с посторонними ионами. Здесь Кт – термодинамическая константа равновесия; аА, аВ, аС и аD – активности исходных веществ А и В и продуктов реакции С и D. Если в изучаемой системе отличия от идеальности обусловлены только электростатическими взаимодействиями А,В,С и Д с посторонними ионами, то состояние равновесия характеризуется реальной (концентрационной) константой 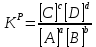 Если посторонние ионы способны вступать с веществами А,В,С и Д в конкурирующие реакции с образованием малодиссоциированных или малорастворимых соединений, то состояние равновесия в такой системе следует характеризовать условной константой равновесия  9. Электролитическая теория Аррениуса. Постулаты 1887 г. - теория Свате Аррениуса (электролитическая) кислотами считаются вещества, при диссоциации которых образуются ионы водорода Н+ (HCl, HNO3), основаниями – вещества, при диссоциации которых образуются гидроксид-ионы ОН- (NaOH, Ca(OH)2). Первый постулат: электролиты обладают способностью при растворении в некоторых растворителях распадаться на противоположно заряженные частицы – ионы. Данный процесс получил название электролитической диссоциации. Различают: бинарные электролиты – 2 иона. Например: одно – одно зарядные (NaCl), двух – двух зарядные (CuSO4). тернарные электролиты – 3 иона. Например: одно-двух зарядные (СuCl2), двух- однозарядные (K2SO4) квартернарные электролиты – 4 иона. Например: одно-трех зарядные (Н3РО4), трех-однозарядные (FeCl3) В 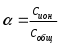 торой постулат: торой постулат:диссоциация идет не полностью. 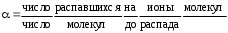 Д  анная величина зависит от: анная величина зависит от:- природы вещества - концентрации в растворе 0 << α < 1 – для сильных электролитов Сион ≈ С 0 < α << 1 для слабых электролитов Сион << С α > 30% сильные электролиты α < 5% слабые электролиты. Третий постулат: силы взаимодействия между ионами отсутствуют. Теория Аррениуса возникла на основе теории растворов Вант-Гоффа, который рассматривал растворы как идеальные газы, и явилась развитием последней отталкиваются  притягиваются притягиваются 10. Константа диссоциации(ионизации). Факторы влияющие на диссоциацию. В  еличина константы ионизации характеризует способность электролита диссоциировать на ионы. Чем больше константа диссоциации, тем больше ионов в его растворе, тем сильнее электролит. Факторы, влияющие на диссоциацию Степень ионизации электролита зависит от его концентрации в растворе. Разбавление раствора ведет к повышению степени диссоциации электролита, потому что с уменьшением его концентрации уменьшается вероятность встречи ионов в растворе. Повышение концентрации электролита в растворе понижает степень его ионизации. Степень диссоциации зависит и от изменения температуры раствора электролита. При повышении температуры степень диссоциации слабого электролита увеличивается, химическая связь в них ослабляется, что облегчает процесс диссоциации электролитов, то есть их распад на ионы. И наоборот, понижение температуры уменьшает степень ионизации слабого электролита. На степень диссоциации влияет добавление одноименных ионов к раствору слабого электролита. Например, если к раствору уксусной кислоты СН3СО-ОН прилить раствор ацетата натрия CH3COONa, то равновесие обратимого процесса диссоциации уксусной кислоты СН3СООН -> СН3СОО- + Н+ согласно принципу Ле-Шателье смещается влево. Поэтому степень диссоциации уксусной кислоты уменьшается. 11. Активность. Коэффициент активности. Ионная сила активности. активная концентрация или активность – эффективной или кажущей концентрации вещества, соответственно которой ионы проявляют себя в химических процессах в качестве реальной действующей массы. Активность связана с равновесной концентрацией простым соотношением. Коэффициент пропорциональности f, называемый коэффициентом активности, характеризует степень отклонения системы от идеальности за счёт электростатических взаимодействий ионов. В идеальной системе аА=[А], так как коэффициент активности равен единице fA=1. Это означает, что электростатические взаимодействия отсутствуют. Для расчёта коэффициентов активности ионов используют теорию Дебая-Хюккеля. Согласно этой теории ион в растворе рассматривается как заряженная частица, окружённая ионной атмосферой. Которая состоит преимущественно из противоположно заряженных ионов, а взаимодействие иона с ионной атмосферой имеет электростатический характер. Величина коэффициента активности зависит от заряда иона и параметров ионной атмосферы: её размеров и плотности. Параметры ионной атмосферы определяются ионной силой раствора I:  12. Теория Дебая-Хюккеля. Приближения. Теория Дебая-Хюккеля и  он в растворе рассматривается как заряженная частица, окружённая ионной атмосферой, которая состоит преимущественно из противоположно заряженных ионов, а взаимодействие иона с ионной атмосферой имеет электростатический характер. Величина коэффициента активности зависит от заряда иона и параметров ионной атмосферы: её размеров и плотности. 13. Теории кислот и оснований, их сопоставительный анализ. В настоящее время наиболее широко используются две теории кислот и оснований: электронная и протонная. В 1923 г. были предложены две, доминирующие по сей день, теории кислот и оснований: протонная теория И. Брёнстеда и Т. Лоури и электронная теория Г. Льюиса. Согласно электронной теории кислот и оснований Льюиса, отличительным признаком кислот и оснований является то, что они взаимодействуют друг с другом с образованием донорно-акцепторной (координационной) связи: А+В → [А:В], По Льюису Определение Кислота - вещество, которое может использовать неподеленную пару электронов атома другой молекулы для образования устойчивой электронной группировки одного из своих атомов, Основание - вещество, обладающее неподеленной парой электронов, которая может быть. использована для образования устойчивой электронной группировки другого атома. По Брёнстеду: Кислота - донор протона водорода, а основание - его акцептор. Протолитическая теория Бренстеда – Лоури. По протолитической, или протонной теории кислота – это соединение, частица которого может отдать протон (Н+) другой частице – основанию. Согласно этой теории кислоты и основания - это вещества, теряющие и приобретающие протоны и называемые протолитами. Передача протона от кислоты к основанию называется протолизом. Кислота–донор протонов,а основание–акцептор Н+. В результате отдачи протона сама кислота превращается в сопряженное ей основание. Основание – акцептор Н+ - превращается в сопряженную ему кислоту. 14. Протолитическая теория Бренстеда–Лоури. Протолитические пары В 1923 году Йоханнес Николаус Бренстед и Томас Мартин Лоури выдвинули положения теории, получившей название протолитической теории кислот и оснований. к кислотам относят те вещества, которые способны отщеплять протоны. основаниями называются вещества, способные присоединять протоны Кислоты и основания – протолиты. Примеры протолитических пар: кислота основание HCl « H+ + Cl- Н2CO3 « H+ + HCO3- HCO3- « H+ + CO3- H2O « H+ + OH- H3O+ « H+ + H2O NH4+ « H+ + NH3 [Zn(OH2)n]2+ « H+ + [Zn(OH)(OH2)n-1]+ [Zn(OH)(H2O)n-1]+ « H+ + [Zn(OH)2(OH2)n-2] Амфипротонные протолиты обладают как протонодонорными, так и протоноакцепторными свойствами.  15. Вывод константа кислотности 16. Вывод константа основности. Т  еория Бренстеда-Лоури учитывает влияние растворителя при диссоциации веществ. То есть процесс диссоциации рассматривается как химическое взаимодействие между растворяемым (диссоциирующим) веществом и растворителем  17.Реакции автопротолиза. Водородный показатель. Большинство растворителей способно проявлять и кислотные, и основные свойства, то есть являются амфотерными (амфипротными). К ним относятся вода, низшие спирты, жидкий аммиак, безводные муравьиная и уксусная кислоты и т.п. Если при столкновении двух молекул амфипротонного растворителя SH одна из них проявляет протонодонорные свойства (кислота), а другая – протоноакцепторные (основание), то между ними протекает протолитическая реакция, называемая реакцией автопротолиза: S  H + SH↔ SH2+ + S- H + SH↔ SH2+ + S-  Вода - слабый электролит Н2О = Н+ + ОН– Ионное произведение воды: Kw = [H+].[OH–] = 10–14 при t=250С Кw не зависит от концентраций ионов Растворы в которых [OH-]=[H+]=10-7 [OH-] > [H+] щелочная среда [OH-] < [H+] кислая среда Характеризовать кислотность и основность растворов концентрацией ионов водорода, выражаемых числами с отрицательными показателями степени, оказалось практически неудобным. Поэтому С.П.Зеренсен предложил реакцию водных растворов характеризовать водородным показателем рН, который равен отрицательному логарифму концентрации [H+]. Т.е. рН = -lg[H+]. Чем меньше величина рН, тем больше концентрация водородных ионов, тем больше кислотность раствора. Наряду с водородным показателем рН применяют гидроксидный показатель рОН. 18.Влияние природы растворителя на силу кислот и оснований. Все вещества лишь потенциально могут быть кислотами или основаниями. Проявить свои кислотные или основные свойства они могут лишь в протолитической реакции. Одним из компонентов такой реакции может быть растворитель. Поэтому существенным достоинством протолитической теории является учет влияния растворителя на процесс кислотно – основного взаимодействия. В зависимости от природы растворителя одно и то же вещество может проявлять как кислотные, так и основные свойства. Например, СН3СООН в воде ведет себя как кислота, отщепляя протон по реакции: СН3СООН + Н2О ->Н3О+ + СН3СОО-, а в безводной серной кислоте - как основание, присоединяя его по реакции: СН3СООН + Н2SO4 -> CH3COOH2+ + HSO4-. Согласно протолитической теории все растворители подразделяются на протолитические и апротонные. 19. Расчет рН растворов сильных кислот.     2   0.Расчет рН растворов слабых кислот. 21.Расчет рН растворов сильных оснований.    22.Расчет рН растворов слабых оснований.   21.Буферные растворы. Механизм буферного действия   Буферные растворы — растворы, сохраняющие постоянное значение pH при добавлении к ним растворов кислот и оснований.  2   3.Определение рН буферных растворов.   24.Вывод константы гидролиза. Гидро́лиз — это химическая реакция взаимодействия вещества с водой, в результате которой происходит разложение этого вещества и воды с образованием новых соединений.   26. Степень гидролиза. Взаимосвязь степени и константы гидролиза. Степень гидролиза — это соотношение количества подвергающейся гидролизу соли nгидр и общего количества растворенной соли nобщ. Обычно, ее обозначают через hгидр (или α ): hгидр = (nгидр/nобщ)·100 % В  еличина hгидр увеличивается с уменьшением силы образующих соль кислоты или основания. 27. Равновесия в системе осадок-раствор. Произведение растворимости и растворимость осадков.   28.Условие выпадения осадка. Влияние различных факторов на растворимость малорастворимых соединений. Условие выпадения осадка Произведение растворимости - важнейшая аналитическая константа, характеризующая основную закономерность равновесного состояния в системе осадок - раствор: в растворе над осадком произведение концентраций ионов является величиной постоянной при данных условиях (температура, растворитель). 1. Если в растворе произведение концентраций ионов или ионное произведение ИП, образующих осадок, меньше произведения растворимости (ИП ПР), раствор насыщен, и осадок не образуется. Молекулы осадка сразу же распадаются на ионы, т.к. их концентрация ниже равновесной. Система стремится к равновесию, и осадок не выпадает. 2. Если ИП ПР, раствор пересыщен, и осадок образуется. Образование осадка будет продолжаться до наступления равенства ИП = ПР и превращения раствора из пересыщенного в насыщенный. Тогда наступает равновесие, и дальнейшее образование осадка прекращается. 3. При равенстве ИП = ПР раствор насыщен, в нем наступает подвижное равновесие, и осадок не выпадает. Влияние факторов При добавлении к насыщенному раствору малорастворимого электролита раствора сильного электролита, не имеющего с ним общих (одноименных) ионов, растворимость малорастворимого электролита увеличивается. Это явление называется солевым эффектом. Влияние растворителя. Растворимость вещества при постоянных температуре и давлении определяется не только свойствами самого вещества, но также свойствами и природой растворителя. |