Лабораторная работа. Нитросоединени я i. Алифатические нитросоединения
 Скачать 191.76 Kb. Скачать 191.76 Kb.
|

Н И Т Р О С О Е Д И Н Е Н И Я I. АЛИФАТИЧЕСКИЕ НИТРОСОЕДИНЕНИЯ Нитросоединения содержат функциональную группу NO2-группы, обладающей очень сильным – М и - I эффектом, и ,во-вторых, образованием стабильного амбидентного аниона, у которого заряд делокализован между атомами кислорода и углерода. 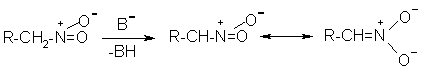 I.1.Получение нитроалканов. Прямое нитрование алканов в жидкой или газовой фазе под действием 50-70%-ной водной азотной кислоты при 500-700оС или тетраоксида азота при 300-500оС имеет промышленное значение только для получения простейших нитроалканов, так как нитрование в этих условиях всегда сопровождается крекингом углеводородов и приводит к сложной смеси самых разнообразных нитросоединений. Эта реакция не получила по этой причине широкого распространения. 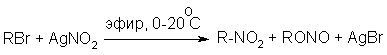 Наиболее общим лабораторным методом получения нитроалканов до сих пор остается реакция алкилирования нитрит-иона, открытая В. Мейером еще в 1872 году. В классическом методе В. Мейера нитрит серебра взаимодействует с первичными или вторичными алкилбромидами и алкилиодидами в эфире, петролейном эфире или без растворителя при 0-20 о С с образованием смеси нитроалкана и алкилнитрита. Нитрит-ион принадлежит к числу вырожденных амбидентных анионов с двумя независимыми нуклеофильными центрами (азот и кислород), которые не связаны в единую мезомерную систему. Реакционная способность амбидентного нитрит-иона с двумя независимыми нуклеофильными центрами (азот и кислород) резко отличается от реакционной способности енолят-ионов с двумя нуклеофильными центрами, связанными в единую мезомерную систему. Соотношение продуктов N- и O- алкилирования (нитроалкан / алкилнитрит) в реакции Мейера алкилбромидов и иодидов с нитритом серебра решающим образом зависит от природы алкильной группы в алкилгалогениде. Выходы первичных нитроалканов достигают 75-85%, однако они резко снижаются до 15-18% для вторичных и до 5% для третичных нитроалканов. Таким образом, ни третичные, ни вторичные алкилгалогениды непригодны для синтеза нитроалканов при взаимодействии с нитритом серебра. Реакция Мейера является по-видимому лучшим способом получения первичных нитроалканов, арилнитрометанов и
Для получения нитроалканов следует использовать только алкилбромиды и алкилиодиды, поскольку алкилхлориды, алкилсульфонаты и диалкилсульфаты не взаимодействуют с нитритом серебра. Из 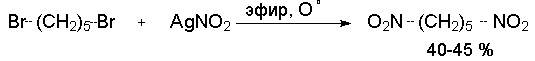 Н. Корнблюм (1955 г.) предложил модифицированный общий метод получения первичных и вторичных нитроалканов, а также Этот метод основан на алкилировании нитритов щелочных металлов первичными или вторичными алкилгалогенидами в диполярном апротонном растворителе ДМФА. Для того чтобы предотвратить последующее нитрозирование нитроалкана параллельно образующимся алкилнитритом, в реакционную смесь необходимо вводить мочевину или многоатомные фенолы - резорцин или флороглюцин. Выход первичных нитроалканов по этому методу не превышает 60 %, т.е. ниже чем при алкилировании нитрита серебра (75-80%). Однако, вторичные нитроалканы можно получать с хорошим выходом алкилированием нитрита натрия в ДМФА. 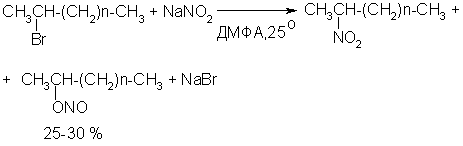 Третичные алкилгалогениды подвергаются элиминированию под действием нитрит-иона и не образуют нитросоединений. Эфиры Другой общий метод синтеза нитроалканов состоит в окислении оксимов кетонов с помощью трифторперуксусной кислоты в ацетонитриле. 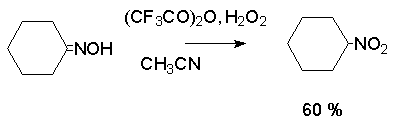 Кроме оксимов можно окислять также и первичные амины перуксусной кислотой или м-хлорпербензойной кислотой : 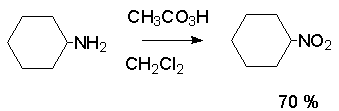 Более ста лет назад Г. Кольбе описал метод получения нитрометана при взаимодействии хлорацетата натрия и нитрита натрия в водном растворе при 80-85oС: 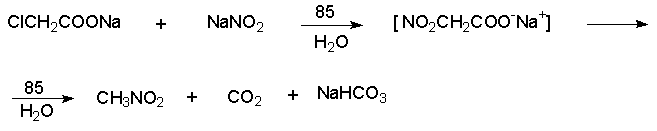 Промежуточно образующийся анион нитроуксусной кислоты декарбоксилируется до нитрометана. Для получения гомологов нитрометана способ Кольбе не имеет значения из-за низкого выхода нитроалканов. Идея этого метода была остроумно использована при разработке современного общего метода получения нитроалканов. Дианионы карбоновых кислот нитруются под действием алкилнитрата с одновременным декарбоксилированием 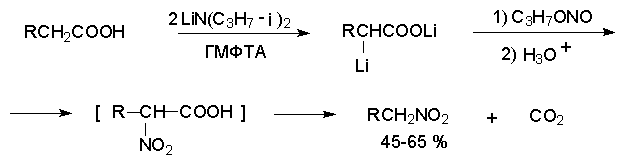 Нитрование карбанионов с помощью алкилнитратов широко используется и для получения 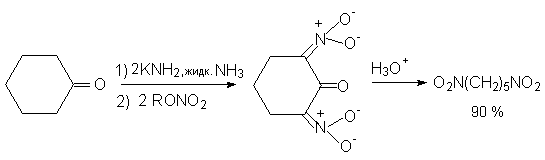 I.2б. Реакции амбидентных анионов нитроалканов. При действии основания как на нитро-форму, так и на аци-форму нитросоединения, образуется общий для них обоих мезомерный амбидентный анион, в котором заряд делокализован между атомами кислорода и углерода. 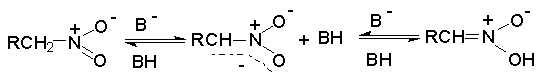 Амбидентные анионы нитроалканов во всех отношениях являются близкими аналогами енолят-ионов карбонильных соединений и для них характерны те же самые реакции замещения, что и для енолят-ионов. Наиболее типичными и важными реакциями с участием анионов нитроалканов являются : галогенирование, алкилирование, ацилирование, конденсации с карбонильными соединениями, реакции Манниха и Михаэля - все те, которые типичны и для енолят-ионов. В зависимости от природы электрофильного агента и в некоторой степени от строения нитроалкана замещение может происходить с участием либо кислородного, либо углеродного, либо обоих центров амбидентного аниона нитроалкана. Галогенирование щелочных солей нитросоединений осуществляется только по атому углерода, реакцию можно остановить на стадии введения одного атома галогена. 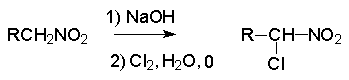 Нитрозирование первичных нитроалканов также осуществляется только по атому углерода и приводит к образованию так называемых нитроловых кислот. 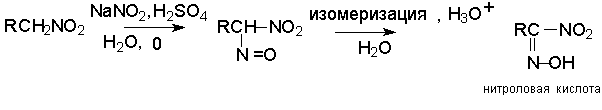 Вторичные нитроалканы в тех же условиях дают псевдонитролы. 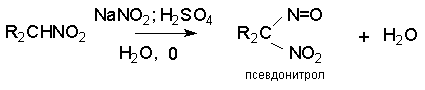 Нитроловые кислоты бесцветны и при встряхивании с раствором гидроксида натрия образуют соли, окрашенные в красный цвет. В отличие от них псевдонитролы обладают в нейтральной среде голубой окраской. Эти соединения могут быть использованы для идентификации первичных и вторичных нитроалканов. Третичные нитроалканы не взаимодействуют при 0о или ниже с азотистой кислотой. Алкилирование амбидентных анионов нитроалканов протекает в отличие от галогенирования и нитрозирования преимущественно по атому кислорода с образованием в качестве промежуточных соединений эфиров аци-формы, которые носят название нитроновых эфиров. Эфиры аци-формы нитроалканов могут быть выделены в индивидуальном виде при алкилировании солей нитроалканов тетрафторборатами триалкилоксония в хлористом метилене при -20о. 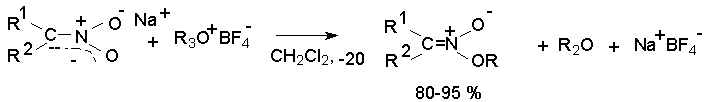 Нитроновые эфиры термически неустойчивы и выше 0-20о подвергаются окислительно-восстановительному распаду на оксим и карбонильное соединение. 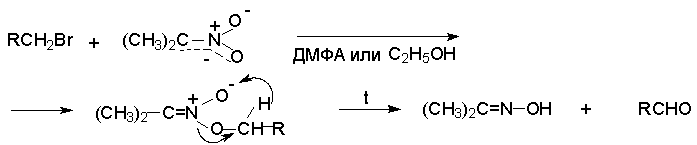 Оксим всегда образуется как конечный продукт восстановления нитроалкана, тогда как альдегид оказывается конечным продуктом окисления алкилирующего агента. Эта реакция нашла широкую область применения в синтезе ароматических альдегидов. При взаимодействии щелочных солей 2-нитропропана с замещенными бензилгалогенидами конечными продуктами являются оксим ацетона и ароматический альдегид. 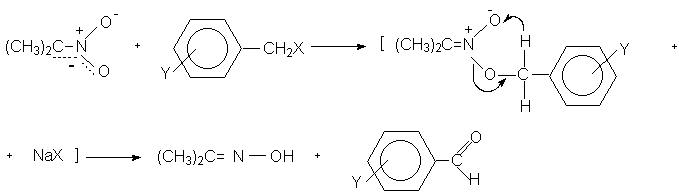 Еще более важную роль играет алкилирование амбидентных анионов нитроалканов под действием аллил-галогенидов для получения 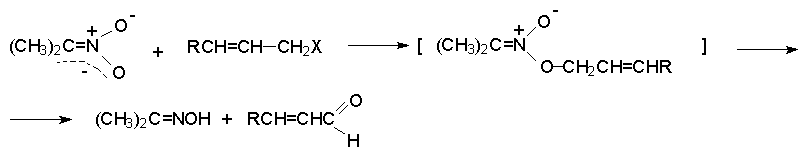 Как следует из приведенных выше примеров, в отличие от енолят-ионов анионы нитроалканов подвергаются региоселективному О-алкилированию. Такое резкое различие в поведении двух родственных классов амбидентных анионов обусловлено высокой степенью локализации заряда на атоме кислорода аниона нитроалкана. При наличии в бензилгалогениде одной или нескольких сильных электроноакцепторных группировок, таких как NO2, NR3, SO2CF3 и др., происходит изменение механизма реакции и ее региоселективности. В этом случае наблюдается С-алкилирование аниона нитроалкана по механизму с участием анион-радикалов, который по своей сути аналогичен SRN1 -механизму ароматического нуклеофильного замещения. 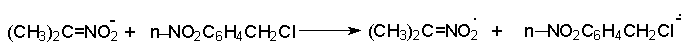 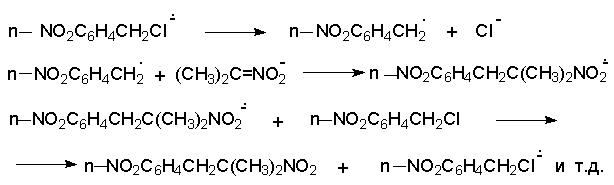 Открытие анион-радикального механизма С-алкилирования нитроалканов и других амбидентных анионов позволило Н.Корнблюму в 1970-1975 годах разработать исключительно эффективный метод алкилирования амбидентных анионов с помощью 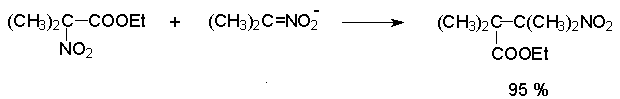 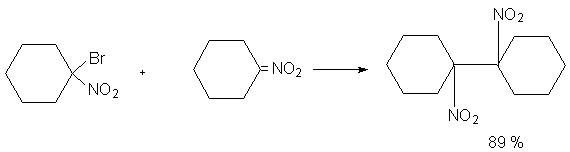 Следует отметить, что в этих реакциях замещение происходит даже у третичного атома углерода. С-Алкилирование удается сделать практически единственным направлением реакции в случае алкилирования дианионов нитроалканов. Дианионы нитроалканов образуются при обработке первичных нитроалканов двумя эквивалентами н.-бутиллития в ТГФ при -100о. 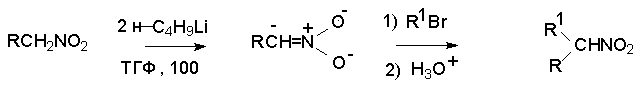 Эти дианионы подвергаются также региоселективному С-ацилированию при взаимодействии с ацилгалогенидами или ангидридами карбоновых кислот. 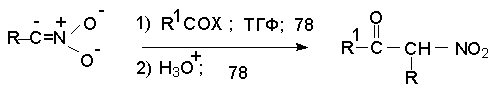 I.2c.Конденсация анионов нитроалканов с карбонильными соединениями (реакция Анри). Конденсация анионов первичных и вторичных нитроалканов с альдегидами и кетонами приводит к образованию 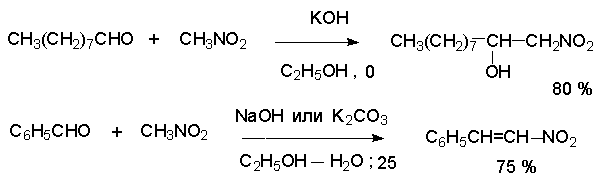 Эта реакция была открыта Л. Анри в 1895 году и может рассматриваться как разновидность альдольно-кротоновой конденсации карбонильных соединений. В конденсации принимает участие анион нитроалкана, а не карбонильного соединения, поскольку кислотность нитроалканов (рКа 10) на десять порядков выше кислотности карбонильных соединений (рКа 20). Эффективными катализаторами реакции Анри являются гидроксиды, алкоксиды и карбонаты щелочных и щелочноземельных металлов. Щелочность среды следует тщательно контролировать для того чтобы исключить альдольную конденсацию карбонильных соединений или реакцию Каниццаро для ароматических альдегидов. Первичные нитроалканы могут также реагировать с двумя молями карбонильного соединения, поэтому соотношение реагентов следует соблюдать очень тщательно. При конденсации ароматических альдегидов обычно образуются только I.2д.Присоединение анионов нитроалкана к активированной двойной связи по Михаэлю и реакция Манниха с участием нитроалканов. Анионы первичных и вторичных нитроалканов присоединяются по кратной связи 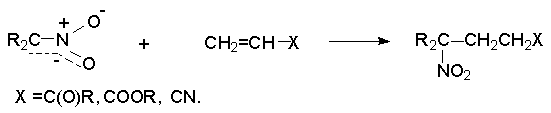 Для первичных нитроалканов реакция может идти и дальше с участием второго моля CH2=CHX. Анионы нитроалканов в реакции присоединения по Михаэлю получают обычным образом с помощью этилата натрия или диэтиламина в качестве основания. 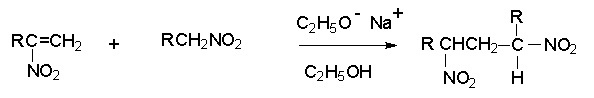 Такого типа присоединение может происходить и в условиях реакции Анри в результате дегидратации продукта конденсации альдегида или кетона с нитроалканом и последующего присоединения нитроалкана. 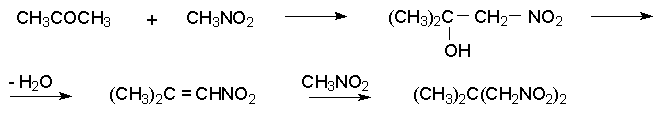 Первичные и вторичные алифатические амины вступают в реакцию Манниха с первичными и вторичными нитроалканами и формальдегидом. 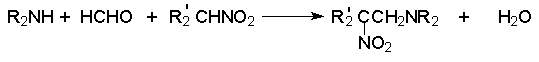 По своему механизму и области применения эта реакция ничем не отличается от классического варианта реакции Манниха с участием карбонильных соединений вместо нитроалканов. II. АРОМАТИЧЕСКИЕ НИТРОСОЕДИНЕНИЯ Методы синтеза и свойства ароматических нитросоединений резко отличаются от описанных выше для нитроалканов. II.1. Получение ароматических нитросоединений. Ароматические нитросоединения получают чаще всего нитрованием аренов, что было подробно рассмотрено при изучении электрофильного ароматического замещения. Другой общий метод получения нитроаренов заключается в окислении первичных ароматических аминов с помощью трифторперуксусной кислоты в хлористом метилене. Трифторперуксусную кислоту получают непосредственно в реакционной смеси при взаимодействии ангидрида трифторуксусной кислоты и 90 %-ной перекиси водорода. Окисление аминогруппы до нитрогруппы с помощью трифторперуксусной кислоты имеет значение для синтеза нитросоединений, содержащих в орто- и пара- положениях другие электроноакцепторные группировки, например, для получения орто- и пара- динитробензола, 1,2,4- тринитробензола, 2,6 - дихлорнитробензола и др.. 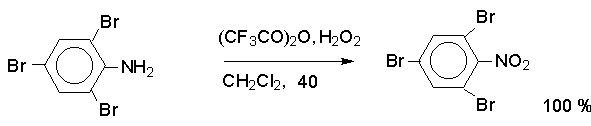 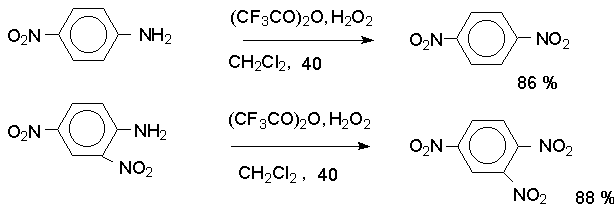 II.2. Свойства ароматических нитросоединений. Нитрогруппа отличается высокой стабильностью по отношению к электрофильным реагентам и разнообразным окислителям. Большинство нуклеофильных агентов за исключением литий- и магнийорганических соединений, а также литийалюминийгидрида не действуют на нитрогруппу. Нитрогруппа относится к числу превосходных нуклеофильных групп в процессах активированного ароматического нуклеофильного замещения ( SNAr). Так, например, нитрогруппа в 1,2,4- тринитробензоле легко замещается под действием гидроксид-, алкоксид-ионов или аминов. 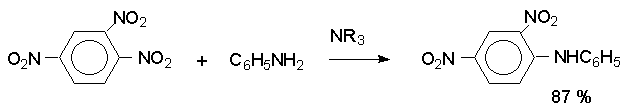 Наиболее важной реакцией ароматических нитросоединений является восстановление их до первичных аминов. Эта реакция была открыта в 1842 году Н.Н.Зининым, который впервые восстановил нитробензол до анилина действием сульфида аммония. В настоящее время для восстановления нитрогруппы в аренах до аминогруппы в промышленных условиях применяется каталитическое гидрирование. В качестве катализатора используют медь на силикагеле в качестве носителя. Катализатор готовят нанесением карбоната меди из суспензии в растворе силиката натрия и последующим восстановлением водородом при нагревании. Выход анилина над этим катализатором составляет 98 %.  Иногда в промышленном гидрировании нитробензола до анилина в качестве катализатора используют никель в комбинации с оксидами ванадия и алюминия. Такой катализатор эффективен в интервале 250-300о и легко регенерируется при окислении воздухом. Выход анилина и других аминов составляет 97-98 %. Восстановление нитросоединений до аминов может сопровождаться гидрированием бензольного кольца. По этой причине для получения ароматических аминов избегают использовать в качестве катализаторов платину. палладий или никель Ренея. Другим методом восстановления нитросоединений является восстановление металлом в кислой или щелочной среде. Восстановление нитрогруппы до аминогруппы происходит в несколько стадий, последовательность которых сильно различается в кислой и щелочной среде. Рассмотрим последовательно процессы, протекающие при восстановлении нитросоединений в кислой и щелочной среде. При восстановлении в кислой среде в качестве восстановителя применяют железо, олово, цинк и соляную кислоту. Эффективным восстановителем нитрогруппы является хлорид олова (II) в соляной кислоте. Этот реагент особенно эффективен в тех случаях, когда в ароматическом нитросоединении есть другие функциональные группы : CHO, COR, COOR и др., чувствительные к действию других восстановителей. Восстановление нитросоединений до первичных аминов в кислой среде происходит ступенчто и включает три стадии с переносом двух электронов на каждой стадии. 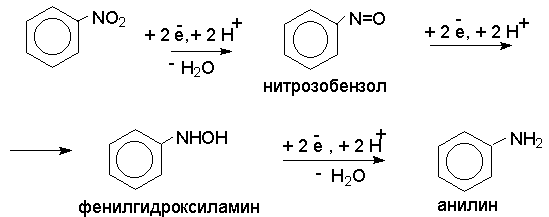 В кислой среде каждый из промежуточных продуктов быстро востанавливается до конечного продукта анилина и их не удается выделить в индивидуальном виде. Однако, в апротонных растворителях в нейтральной среде можно зафиксировать промежуточные продукты восстановления. При восстановлении нитробензола натрием или калием в ТГФ сначала образуется анион-радикал нитробензола за счет переноса одного электрона от щелочного металла. 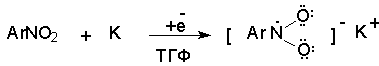 Катион щелочного металла связан в контактную ионную пару с атомом кислорода нитрогруппы анион-радикала. При дальнейшем восстановлении анион-радикал превращается в дианион, который после протонирования дает нитрозобензол. 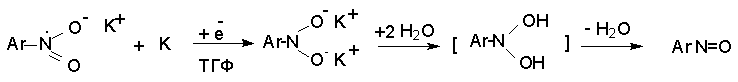 Нитрозобензол, также как и другие ароматические нитрозосоединения, обладает высоким окислительным потенциалом и очень быстро восстанавливается до N-фенилгидроксиламина. Поэтому нитрозобензол не удается выделить в качестве промежуточного продукта восстановления, хотя данные электрохимического восстановления однозначно указывают на его образование. Дальнейшее восстановление нитрозосоединений до N-арилгидроксиламина включает две аналогичные стадии одноэлектронного восстановления до анион-радикала и далее до дианиона нитрозосоединения, который при протонировании превращается в N-арилгидроксиламин. 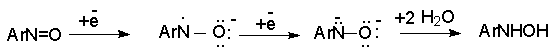 Последняя стадия восстановления арилгидроксиламина до первичного амина сопровождается гетеролитическим расщеплением связи азот-кислород после протонирования субстрата. В нейтральном водном растворе можно получить фенилгидроксиламин в качестве продукта восстановления нитробензола. Фенилгидроксиламин получается при восстановлении нитробензола цинком в водном растворе хлорида аммония. 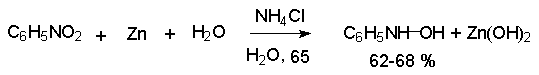 Арилгидроксиламины легко восстанавливаются в амины при обработке железом или цинком и соляной кислотой. 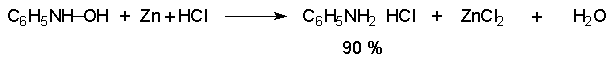 Поскольку фенилгидроксиламин является промежуточным продуктом восстановления, его можно не только восстановить до анилина, но и окислить до нитрозобензола. 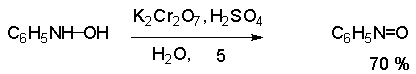 Это, вероятно, один из лучших методов получения ароматических нитрозосоединений, которые не удается иным способом выделить в качестве промежуточного продукта восстановления нитросоединений. Ароматические нитрозосоединения легко димеризуются в твердом состоянии, причем их димеры бесцветны. В жидком и газообразном состоянии они мономерны и окрашены в зеленый цвет. 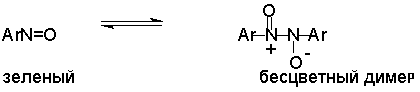 Восстановление нитросоединений металлами в щелочной среде отличается от восстановления в кислой среде. В щелочной среде нитрозобензол быстро взаимодействует со вторым промежуточным продуктом восстановления фенилгидроксиламином с образованием азоксибензола. Эта реакция по существу подобна присоединению азотистых оснований к карбонильной группе альдегидов и кетонов. 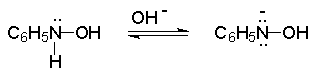 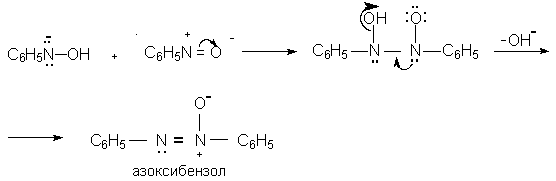 В лабораторных условиях азоксибензол с хорошим выходом получается при восстановлении нитросоединений боргидридом натрия в ДМСО, метилатом натрия в метиловом спирте или старым способом при использовании в качестве восстановителя As2O3 или глюкозы. 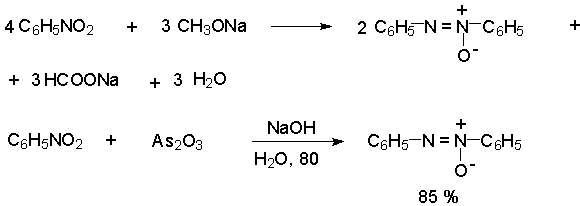 Азоксибензол при действии цинка в спиртовом растворе щелочи восстанавливается сначала до азобензола, а при действии избытка цинка далее до гидразобензола. 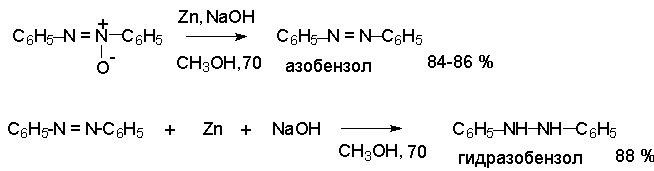 В синтетической практике производные азоксибензола могут быть восстановлены до азобензола под действием триалкилфосфита в качестве восстановителя. С другой стороны, азобензол легко окисляется до азоксибензола перкислотами. 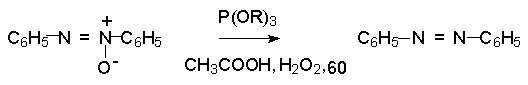 Азобензол существует в виде цис- и транс- изомеров. При восстановлении азоксибензола получается более стабильный транс-изомер, который при облучении УФ-светом превращается в цис-изомер. 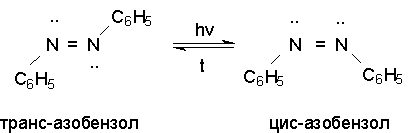 Несимметричные производные азобензола получаются при конденсации нитрозосоединений и первичных ароматических аминов. При восстановлении ароматических нитросоединений алюмогидридом лития в эфире также образуются азосоединения с выходом, близким к количественному. 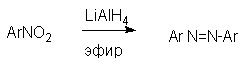 Азобензол восстанавливается цинковой пылью и спиртовой щелочью до гидразобензола. Гидразобензол является, таким образом, конечным продуктом восстановления нитробензола металлом в щелочной среде. На воздухе бесцветный гидразобензол легко окисляется до окрашенного в оранжево-красный цвет азобензола. Вместе с тем гидразобензол, также как и азобензол и азоксибензол, восстанавливается до анилина под действием дитионита натрия в воде или хлорида олова (II) в соляной кислоте. Суммарный процесс восстановления ароматических нитросоединений металлами в кислой и щелочной среде может быть представлен в виде следующей последовательности превращений. В кислой среде : В щелочной среде: 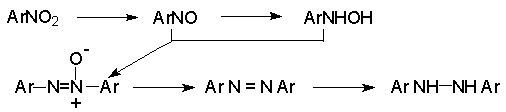 В промышленности анилин получают каталитическим восстановлением нитробензола на медном или никелевом катализаторе, который вытеснил старинный способ восстановления нитробензола чугунными стружками в водном растворе хлорного железа и соляной кислоты. Восстановление нитрогруппы до аминогруппы сульфидом и гидросульфидом натрия в настоящее время имеет значение только для частичного восстановления одной из двух нитрогрупп, например, в м-динитробензоле или 2,4-динитроанилине. 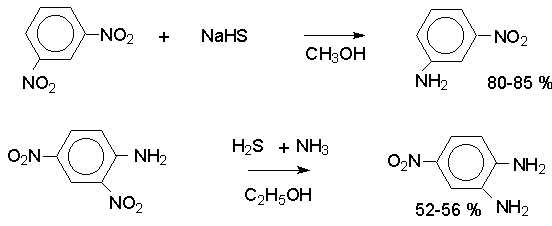 При ступенчатом восстановлении полинитросоединений с помощью сульфида натрия этот неорганический реагент превращается в тетрасульфид натрия, что сопровождается образованием щелочи. Высокая щелочность среды приводит к образованию азокси- и азосоединений в качестве побочных продуктов. Для того чтобы избежать этого в качестве восстановителя следует использовать гиросульфид натрия, где щелочь не образуется. II.3. Бензидиновая перегруппировка. Превращение гидразобензола в 4,41 - диаминобифенил, названный бензидином, под действием сильных минеральных кислот было открыто в 1845 году Н.Н.Зининым. Это превращение впоследствие получило название бензидиновой перегруппировки. В настоящее время этим термином объединяют целую группу родственных перегруппировок, приводящих к образованию смеси орто- и пара-изомерных производных диаминобифенила. При перегруппировке самого гидразобензола получается смесь диаминов, содержащая 70 % бензидина и 30 % 2,41-диаминобифенила. 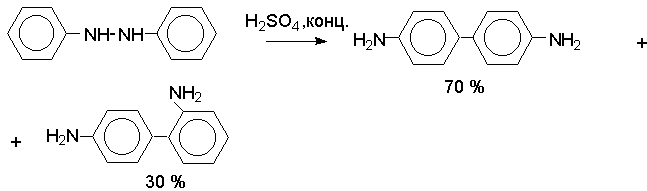 Если пара-положение в одном из бензольных ядер гидразобензола занято каким-нибудь заместителем, продуктом перегруппировки оказывается производное дифениламина ( так называемая семидиновая перегруппировка). 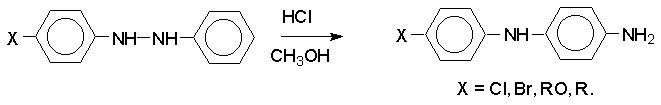 При изучении механизма бензидиновой и родственных перегруппировок было установлено, что они протекают внутримолекулярно. Если два различных гидразобензола подвергнуть совместной перегруппировке, то продукты перекрестной перегруппировки отсутствуют. Для перегруппировки самого гидразобензола было обнаружено, что скорость реакции пропорциональна концентрации гидразобензола и квадрату концентрации протона. Это означает, что перегруппировке подвергается дипротонированная форма гидразобензола. Было также показано, что монопротонированная форма гидразобензола превращается нацело в бензидин только при повторной обработке кислотой. Эти данные согласуются со следующим механизмом бензидиновой перегруппировки. 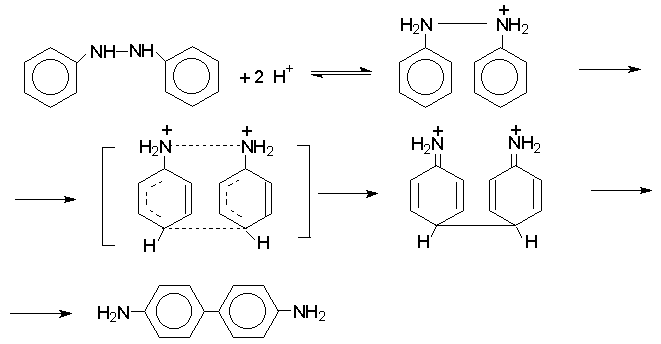 Предполагается. что переходное состояние образуется из такой конформации гидразобензола, в которой сильно сближены между собой два соответствующих атома углерода обоих бензольных колец. Согласно квантовохимическому расчету расстояние между взаимодействующими центрами в такой конформации составляет всего 0,15 нм и почти не отличается от длины центральной углерод-углеродной связи двух колец бифенила. Образование новой углерод-углеродной связи и разрыв старой связи двух атомов азота происходит строго синхронно. Согласно современной терминологии бензидиновая перегруппировка относится к числу [5,5] - сигматропных перегруппировок. Бензидин и его производные канцерогенны и вызывают рак мочевого пузыря. По этой причине производство бензидина из года в год резко сокращается. Использование бензидина в текстильной промышленности запрещено. |