Общие рекомендации
 Скачать 4.72 Mb. Скачать 4.72 Mb.
|

Основные положения хромосомной теории наследственностиАнализ явлений сцепленного наследования, кроссинговера, сравнение генетической и цитологической карт позволяют сформулировать основные положения хромосомной теории наследственности:
Сцепленное наследованиеНезависимое комбинирование признаков (третий закон Менделя) осуществляется при условии, что гены, определяющие эти признаки, находятся в разных парах гомологичных хромосом. Следовательно, у каждого организма число генов, способных независимо комбинироваться в мейозе, ограничено числом хромосом. Однако в организме число генов значительно превышает количество хромосом. Например, у кукурузы до эры молекулярной биологии было изучено более 500 генов, у мухи дрозофилы — более 1 тыс., а у человека — около 2 тыс. генов, тогда как хромосом у них 10, 4 и 23 пары соответственно. То, что число генов у высших организмов составляет несколько тысяч, было ясно уже У. Сэттону в начале XX века. Это дало основание предположить, что в каждой хромосоме локализовано множество генов. Гены, локализованные в одной хромосоме, образуют группу сцепления и наследуются вместе. Совместное наследование генов Т. Морган предложил назвать сцепленным наследованием. Число групп сцепления соответствует гаплоидному числу хромосом, поскольку группу сцепления составляют две гомологичные хромосомы, в которых локализованы одинаковые гены. (У особей гетерогаметного пола, например, у самцов млекопитающих, групп сцепления на самом деле на одну больше, так как X- и У-хромосомы содержат разные гены и представляют собой две разные группы сцепления. Таким образом, у женщин 23 группы сцепления, а у мужчин — 24). Способ наследования сцепленных генов отличается от наследования генов, локализованных в разных парах гомологичных хромосом. Так, если при независимом комбинировании дигетерозиготная особь образует четыре типа гамет (АВ, Ab, аВ и ab) в равных количествах, то при сцепленном наследовании (в отсутствие кроссинговера) такая же дигетерозигота образует только два типа гамет: (АВ и ab) тоже в равных количествах. Последние повторяют комбинацию генов в хромосоме родителя. Было установлено, однако, что кроме обычных (некроссоверных) гамет возникают и другие (кроссоверные) гаметы с новыми комбинациями генов — Ab и аВ, отличающимися от комбинаций генов в хромосомах родителя. Причиной возникновения таких гамет является обмен участками гомологичных хромосом, или кроссинговер. Кроссинговер происходит в профазе I мейоза во время конъюгации гомологичных хромосом. В это время части двух хромосом могут перекрещиваться и обмениваться своими участками. В результате возникают качественно новые хромосомы, содержащие участки (гены) как материнских, так и отцовских хромосом. Особи, которые получаются из таких гамет с новым сочетанием аллелей, получили название кроссинговерных или рекомбинантных. Частота (процент) перекреста между двумя генами, расположенными в одной хромосоме, пропорциональна расстоянию между ними. Кроссинговер между двумя генами происходит тем реже, чем ближе друг к другу они расположены. По мере увеличения расстояния между генами все более возрастает вероятность того, что кроссинговер разведет их по двум разным гомологичным хромосомам. Расстояние между генами характеризует силу их сцепления. Имеются гены с высоким процентом сцепления и такие, где сцепление почти не обнаруживается. Однако при сцепленном наследовании максимальная частота кроссинговера не превышает 50 %. Если же она выше, то наблюдается свободное комбинирование между парами аллелей, не отличимое от независимого наследования. Биологическое значение кроссинговера чрезвычайно велико, поскольку генетическая рекомбинация позволяет создавать новые, ранее не существовавшие комбинации генов и тем самым повышать наследственную изменчивость, которая дает широкие возможности адаптации организма в различных условиях среды. Человек специально проводит гибридизацию с целью получения необходимых вариантов комбинаций для использования в селекционной работе. Кроссинговер. Этот процесс происходит в профазе I мейоза в то время, когда гомологичные хромосомы тесно сближены в результате конъюгации и образуют биваленты. В ходе кроссинговера осуществляется обмен соответствующими участками между взаимно переплетающимися хроматидами гомологичных хромосом (рис. 3.72). Этот процесс обеспечивает перекомбинацию отцовских и материнских аллелей генов в каждой группе сцепления. В разных предшественниках гамет Кроссинговер происходит в различных участках хромосом, в результате чего образуется большое разнообразие сочетаний родительских аллелей в хромосомах. 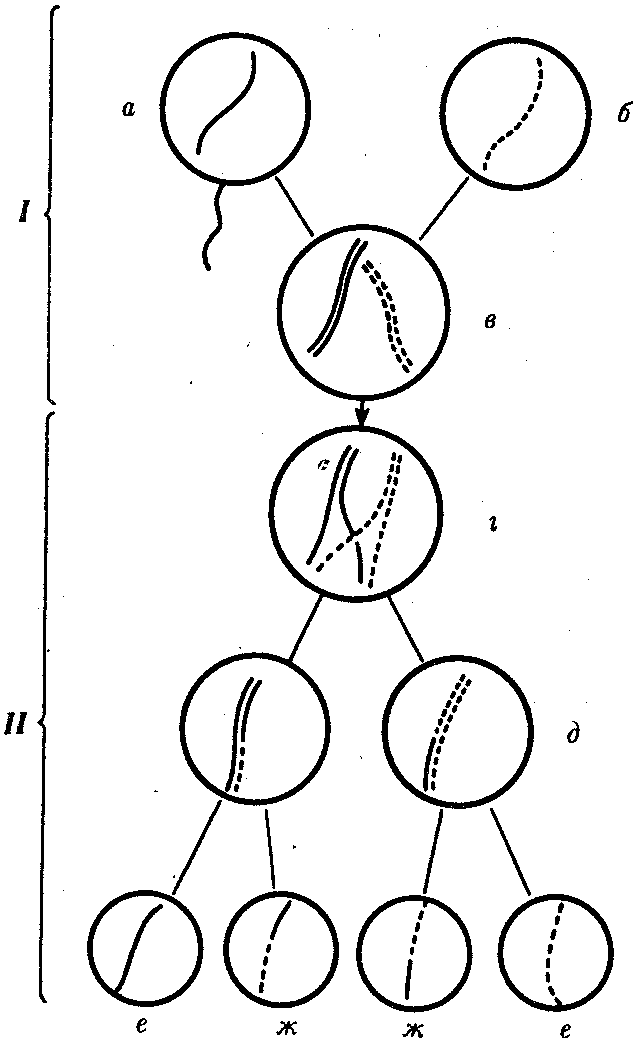 Рис. 3.72. Кроссинговер как источник генетического разнообразия гамет: I — оплодотворение родительских гамет а и б с образованием зиготы в; II — гаметогенез в организме, развившемся из зиготы в; г — кроссинговер, происходящий между гомологами в профазе I;д — клетки, образовавшиеся после 1-го мейотического деления; е, ж — клетки, образовавшиеся после 2-го деления мейоза (е — некроссоверные гаметы с исходными родительскими хромосомами; ж — кроссоверные гаметы с перекомбинацией наследственного материала в гомологичных хромосомах) Понятно, что кроссинговер как механизм рекомбинации эффективен лишь в том случае, когда соответствующие гены отцовской и материнской хромосом представлены разными аллелями. Абсолютно идентичные группы сцепления при кроссинговере не дают новых сочетаний аллелей. Кроссинговер происходит не только в предшественницах половых клеток при мейозе. Он наблюдается также в соматических клетках при митозе. Соматический кроссинговер описан у дрозофилы, у некоторых видов плесеней. Он осуществляется в ходе митоза между гомологичными хромосомами, однако его частота в 10 000 раз меньше частоты мейотического кроссинговера, от механизма которого он ничем не отличается. В результате митотического кроссинговера появляются клоны соматических клеток, различающихся по содержанию в них аллелей отдельных генов. Если в генотипе зиготы данный ген представлен двумя разными аллелями, то в результате соматического кроссинговера могут появиться клетки с одинаковыми либо отцовскими, либо материнскими аллелями данного гена (рис. 3.73). 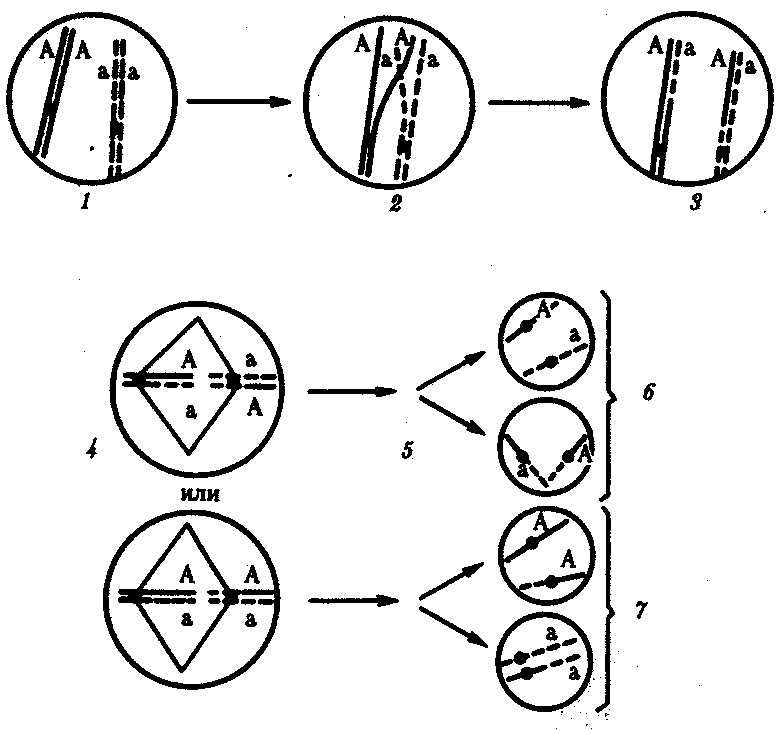 Рис. 3.73. Кроссинговер в соматических клетках: 1 — соматическая клетка, в гомологичных хромосомах которой ген А представлен двумя разными аллелями (А и а); 2 — кроссинговер; 3 — результат обмена соответствующими участками между гомологичяыми хромосомами; 4 — расположение гомологов в плоскости экватора веретена деления в метафазе митоза (два варианта); 5 — образование дочерних клеток; 6 — образование гетерозитотиых по гену А клеток, сходных с материнской клеткой по набору аллелей (Аа); 7 — образование гомозиготных по гену А клеток, отличающихся от материнской клетки по набору аллелей (АА или аа) 40.Наследование. Типы наследования. Особенности аутосомного, Х-сцепленного и голандрического типов наследования. Полигенное наследование. под наследственностью понимают свойство клеток или организмов в процессе самовоспроизведения передавать новому поколению способность к определенному типу обмена веществ и индивидуального развития, в ходе которого у них формируются общие признаки и свойства данного типа клеток и вида организмов, а также некоторые индивидуальные особенности родителей. На популяционно-видовом уровне организации жизни наследственность проявляется в поддержании постоянного соотношения различных генетических форм в ряду поколений организмов данной популяции (вида). Наследственность – свойство живых организмов, обеспечивающее материальную преемственность онтогенеза в определенных условиях внешней среды. Гены детерминируют последовательность полипептидной цепи. Наследование – передача информации от одного поколения к другому. Благодаря наследственности стало возможно существование популяций, видов и других групп.  Аутосомное наследование. Характерные черты аутосомного наследования признаков обусловлены тем, что соответствующие гены, расположенные в аутосомах, представлены у всех особей вида в двойном наборе. Это означает, что любой организм получает такие гены от обоих родителей. В соответствии с законом чистоты гамет в ходе гаметогенеза все половые клетки получают по одному гену из каждой аллельной пары (рис. 6.6). Обоснованием этого закона является расхождение гомологичных хромосом, в которых располагаются аллельные гены, к разным полюсам клетки в анафазе I мейоза Ввиду того что развитие признака у особи зависит в первую очередь от взаимодействия аллельных генов, разные его варианты, определяемые разными аллелями соответствующего гена, могут наследоваться по аутосомно-доминантному или аутосомно-рецессивному типу, если имеет место доминирование. Возможен также промежуточный тип наследования признаков при других видах взаимодействия аллелей (см. разд. 3.6.5.2). При доминировании признака, описанном Г. Менделем в его опытах на горохе, потомки от скрещивания двух гомозиготных родителей, различающихся по доминантному и рецессивному вариантам данного признака, одинаковы и похожи на одного из них (закон единообразия F1). Описанное Менделем расщепление по фенотипу в F2 в отношении 3:1 в действительности имеет место лишь при полном доминировании одного аллеля над другим, когда гетерозиготы фенотипически сходны с доминантными гомозиготами (закон расщепления в F2). Наследование рецессивного варианта признака характеризуется тем, что он не проявляется у гибридов F1, а в F2 проявляется у четверти потомков. В случаях формирования у гетерозигот нового варианта признака по сравнению с гомозиготами, что наблюдается при таких видах взаимодействия аллельных генов, как неполное доминирование, кодоминирование, межаллельная комплементация, гибриды F1 не похожи на родителей, а в F2 образуется три фенотипических группы потомков (рис. 6.7, II). Сцепленное с полом наследование. Х-сцепленное наследование. Х-хромосома присутствует в кариотипе каждой особи, поэтому признаки, определяемые генами этой хромосомы, формируются у представителей как женского, так и мужского пола. Особи гомогаметного пола получают эти гены от обоих родителей и через свои гаметы передают их всем потомкам. Представители гетерогаметного пола получают единственную Х-хромосому от гомогаметного родителя и передают ее своему гомогаметному потомству. У млекопитающих (в том числе и человека) мужской пол получает Х-сцепленные гены от матери и передает их дочерям. При этом мужской пол никогда не наследует отцовского Х-сцепленного признака и не передает его своим сыновьям. Так как у гомогаметного пола признак развивается в результате взаимодействия аллельных генов, различают Х-сцепленное доминантное и Х-сцепленное рецессивное наследование. Х-сцепленный доминантный признак (красный цвет глаз у дрозофилы) передается самкой всему потомству. Самец передает свой Х-сцепленный доминантный признак лишь самкам следующего поколения. Самки могут наследовать такой признак от обоих родителей, а самцы — только от матери. Х-сцепленный рецессивный признак, (белый цвет глаз у дрозофилы) у самок проявляется только при получении ими соответствующего аллеля от обоих родителей (XaXa). У самцов XaY он развивается при получении рецессивного аллеля от матери. Рецессивные самки передают рецессивный аллель потомкам любого пола, а рецессивные самцы —только «дочерям» (см. рис. 6.8). Голандрическое наследование. Активно функционирующие гены Y-хромосомы, не имеющие аллелей в Х-хромосоме, присутствуют в генотипе только гетерогаметного пола, причем в гемизиготном состоянии. Поэтому они проявляются фенотипически и передаются из поколения в поколение лишь у представителей гетерогаметного пола. Так, у человека признак гипертрихоза ушной раковины («волосатые уши») наблюдается исключительно у мужчин и наследуется от отца к сыну. Большинство количественных признаков организмов определяется полигенами, т.е. системой неаллельных генов, одинаково влияющих на формирование данного признака. Взаимодействие таких генов в процессе формирования признака называют полимерным. Оно сводится чаще всего к суммированию действия сходных аллелей этих генов, определяющих формирование одинакового варианта признака. Совместное действие полигенов обусловливает различную экспрессивность — степень выраженности признака, зависящую от дозы соответствующих аллелей. В основе появления в геноме таких генов, очевидно, лежит их дупликация или амплификация (см. разд. 3.6.4.3). Это позволяет увеличить синтез соответствующего продукта в клетках организма. По полимерному типу взаимодействия у человека определяется интенсивность окраски кожных покровов, зависящая от уровня отложения в клетках пигмента меланина. В геноме человека имеется четыре гена, отвечающих за этот признак. В генотипе все они представлены в двойной дозе. В связи с тем что эти гены отвечают за развитие одного и того же признака, их обозначают одной и той же буквой, но с разными символами, чтобы подчеркнуть их неаллельность — P1P2P3P4. Наличие в генотипе восьми доминантных аллелей в системе полигенов, определяющих цвет кожи, обусловливает максимальную ее пигментацию, наблюдаемую у африканских негров (P1P1P2P2P3P3P4P4). Полное отсутствие доминантных аллелей у. рецессивных гомозигот (р1р2р2р3р3р4р4) проявляется в виде минимальной пигментации у европеоидов. Большее или меньшее количество доминантных аллелей, колеблющееся от 8 до 0, обеспечивает разную интенсивность окраски кожи (рис. 3.80). Полимерное взаимодействие генов лежит в основе определения главным образом количественных признаков (рост, масса организма, возможно, интеллект). 41.Количественная и качественная специфика проявления генов в признаках: пенетрантность, экспрессивность, плейотропность, генокопии. Никакие признаки не наследуются. Признаки развиваются на основе взаимодействия генотипа и среды. Наследуется только генотип, т.е. комплекс генов, который определяет норму биологической реакции организма, изменяющую проявление и выраженность признаков в разных условиях среды. Таким образом, организм реагирует на свойства внешней среды. Иногда один и тот же ген в зависимости от генотипа и от условий внешней среды по-разному проявляет признак или меняет полноту выраженности. Степень проявления фенотипа – экспрессивность. Образно ее можно сравнить со степенью тяжести болезни в клинической практике. Экспрессивность подчиняется законам распределения Гаусса (некоторые в малом или среднем количестве). В основе изменчивости экспрессивности лежат и генетические факторы, и факторы внешней среды. Экспрессивность – очень важный показатель фенотипического проявления гена. Количественно ее степень определяют, используя статистический показатель. Экспрессивность также является показателем, характеризующим фенотипическое проявление наследственной информации. Она характеризует степень выраженности признака и, с одной стороны, зависит от дозы соответствующего аллеля гена при моногенном наследовании или от суммарной дозы доминантных аллелей генов при полигенном наследовании, а с другой — от факторов среды. Примером служит интенсивность красной окраски цветков ночной красавицы, убывающая в ряду генотипов АА, Аа, аа, или интенсивность пигментации кожи у человека, увеличивающаяся при возрастании числа доминантных аллелей в системе полигенов от 0 до 8 (см. рис. 3.80). Влияние средовых факторов на экспрессивность признака демонстрируется усилением степени пигментации кожи у человека при ультрафиолетовом облучении, когда появляется загар, или увеличением густоты шерсти у некоторых животных в зависимости от изменения температурного режима в разные сезоны года. Генетический признак может даже не проявляться в некоторых случаях. Если ген есть в генотипе, но он вовсе не проявляется – он пенетрирован. (русский ученый Тимофеев-Рисовский 1927 год). Пенетрантность – количество особей (%), проявляющих в фенотипе данный ген, по отношению к количеству особей, у которых этот признак мог бы проявиться. Пенетрантность свойственна проявлению многих генов. Важен принцип – «все или ничего» - либо проявляется, любо нет. - наследственный панкреатит – 80% - вывих бедра – 25% - пороки развития глаз - ретинобластома – 80% - отосклероз – 40%
Хорея Гентингтона проявляется в непроизвольном подергивании головы. Конечностей, постепенно прогрессирует и приводит к смерти. Может проявиться в раннем постэмбриональном периоде, в зрелом возрасте или не проявиться вообще. И экспрессивность, и пенетрантность поддерживаются естественным отбором, т.е. гены, контролирующие патологические признаки могут иметь разную экспрессивность и пенетрантность: заболевают не все носители гена, а у заболевших степень проявления будет различна. Проявление или неполное проявление признака, а так же его отсутствие зависит от среды и от модифицирующего действия других генов. Ген может действовать плейотропно (множественно), т.е. опосредовано влиять на течение разных реакций и развитие многих признаков. Гены могут оказывать влияние на другие признаки на разных стадиях онтогенеза. Если ген включается в позднем онтогенезе, то оказывается незначительное действие. Если на ранних стадиях – изменения более значительны. Фенилкетанурия. У больных есть мутация, которая выключает фермент – фенилаланин – гидролазу. Поэтому фенилаланин не превращается в тирозин. В результате в крови количество фенилаланина повышается. Если выявить эту патологию рано (до 1 месяца) и перевести ребенка на другое питание, развитие идет нормально, если позднее – понижен размер головного мозга, умственная отсталость, не развиваются нормально, отсутствует пигментация, умственные способности минимальны. Плейотропность отражает интеграцию генов и признаков. У человека есть патологический ген, приводящий к синдрому Фанкони (порок развития или отсутствие большого пальца, порок или отсутствие лучевой кости, недоразвитие почки, коричневые пигментные пятна, нехватка кровяных телец). Есть ген, связанный с Х-хромосомой. Невосприимчивость к инфекциям и нехватка кровяных телец. Доминантный ген, сцепленный с Х-хромосомой – пилонефрит, лабиринтная тугоухость. Синдром Марфани – паучьи пальцы, вывих хрусталика глаза, пороки развития сердца. Генокопия (греч. genos род, происхождение + лат. copia множество) - термин в 1957 г. предложил немецкий генетик Нахтсхейм (H.Nachtsheim).Обозначает сходные изменения одного и того же признака под влиянием разных неаллельных генов, которые иногда называют миметическими генами гетерогенной группы. genocopies - генокопии. Oдинаковые изменения фенотипа, обусловленные аллелями разных генов, а также имеющие место в результате различных генных взаимодействий или нарушений различных этапов одного биохимического процесса с прекращением синтеза конечного продукта, - например, у Drosophila melanogaster известен ряд мутаций неаллельных генов, обусловливающих фенотип “красные глаза” (нарушен синтез коричневого пигмента). 42.Изменчивость. Формы изменчивости: модификационная и генотипическая, их значение в онтогенезе и эволюции. Изменчивость Изменчивость – свойство живых организмов существовать в разных формах. Групповая и индивидуальная изменчивость – классификация по эволюционному значению. Изменчивость, реализованная группой организмов, называется групповой, у одного организма или группы его клеток – индивидуальная. По характеру изменения признаков и механизму: --фенотипическая - случайная - модификационная --генотипическая - соматическая - генеративная (мутационная, комбинативная) а) генная б) хромосомная в) геномная Модификационная изменчивость отражает изменение фенотипа под воздействием факторов внешней среды (усиление и развитие мышечной и костной массы у спортсменов, увеличение эритропоэза в условиях высокогорья и крайнего севера). 43.Фенотипическая изменчивость и её виды. Модификации и их характеристики. Норма реакции признака. Фенокопии. Адаптивный характер модификаций. По характеру изменения признаков и механизму: --фенотипическая - случайная - модификационная Модификационная изменчивость отражает изменение фенотипа под воздействием факторов внешней среды (усиление и развитие мышечной и костной массы у спортсменов, увеличение эритропоэза в условиях высокогорья и крайнего севера). Частный случай фенотипической изменчивости – фенокопии. Фенокопии – вызванные условиями внешней среды фенотипические модификации, имитирующие генетические признаки. Под влиянием внешних условий на генетически нормальный организм копируются признаки совсем другого генотипа. Проявление дальтонизма может произойти под влиянием питания, плохой психической конституции, повышенной раздражительности. У человека возникает заболевание витилиго (1% людей) – нарушение пигментации кожи. Генетический дефект есть у 30% болеющих, у остальных – профессиональное витилиго (воздействие на организм особых химических и отравляющих веществ). В Германии 15 лет назад рождались дети с фекомелией – укороченными ластовидными руками. Выяснилось. Что рождение таких детей происходило, если мать принимала Телидомид (успокоительное средство, показанное беременным). В результате нормальный немутантный генотип получал мутацию. Фенокопии появляются в большинстве случаев при действии внешней среды на ранних стадиях эмбриогенеза, что приводит к врожденным заболеваниями порокам развития. Наличие фенокопий затрудняет диагностику заболеваний. Норма реакцииПредел проявления модификационной изменчивости организма при неизменном генотипе — норма реакции. Норма реакции обусловлена генотипом и различается у разных особей данного вида. Фактически норма реакции — спектр возможных уровней экспрессии генов, из которого выбирается уровень экспрессии, наиболее подходящий для данных условий окружающей среды. Норма реакции имеет предел для каждого вида — например, усиленное кормление приведет к увеличению массы животного, однако она будет находиться в пределах нормы реакции, характерной для данного вида или породы. Норма реакции генетически детерминирована и наследуется. Для разных изменений есть разные пределы нормы реакции. Например, сильно варьируют величина удоя, продуктивность злаков (количественные изменения), слабо — интенсивность окраски животных и т. д. (качественные изменения). В соответствии с этим норма реакции может быть широкой (количественные изменения — размеры листьев многих растений, размеры тела многих насекомых в зависимости от условий питания их личинок) и узкой (качественные изменения — окраска у куколок и имаго некоторых бабочек). Тем не менее, для некоторых количественных признаков характерна узкая норма реакции (жирность молока, число пальцев на ногах у морских свинок), а для некоторых качественных признаков — широкая (например, сезонные изменения окраски у многих видов животных северных широт). Фенотипические изменения, возникающие на основе одного и того же генотипа в разных условиях его реализации, называют модификациями. Примером модификаций могут служить изменения содержания жира в молоке животных или массы тела в зависимости от их питания, изменения количества эритроцитов в крови, в зависимости от парциального давления кислорода в воздухе, изменения темпа роста растений при разной освещенности и содержании минеральных веществ в почве. Другим примером модификационной изменчивости являются различия, наблюдаемые у генетически идентичных монозиготных близнецов или потомков одного растения, полученных путем вегетативного размножения, но развивавшихся в разных условиях среды. Модификации отдельного признака или свойства, формируемого данным генотипом, образуют непрерывный ряд. Частота встречаемости каждого варианта в таком вариационном ряду различна. Чаще обнаруживаются средние значения признака. Чем дальше признак отстоит от среднего значения, тем реже он наблюдается (рис. 6.1). Так как фенотипическое проявление наследственной информации может модифицироваться условиями среды, в генотипе организма запрограммировано не конкретное значение отдельных его характеристик, а лишь возможность их формирования в определенных пределах, называемых нормой реакции. Таким образом, норма реакции представляет собой пределы модификационной изменчивости признака, допустимой при данном генотипе. Некоторые признаки характеризуются широкой нормой реакции. Как правило, это количественные признаки, контролируемые полигенами (масса тела, жирность молока, пигментация кожи), другие свойства характеризуются узкой нормой реакции и слабо или почти не модифицируются в разных условиях (цвет глаз, группа крови). 44.Фенотип. Фенотип как результат реализации наследственной информации (генотипа) в определенных условиях среды. Значение средовых и генотипических факторов в формировании патологически измененного фенотипа человека. 45.Комбинативная изменчивость, её механизмы. Значение комбинативной изменчивости в обеспечении генотипического разнообразия людей. 46.Генные болезни человека, механизмы их возникновения и проявления. Примеры. Генные болезни – это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена. В зависимости от функциональной значимости первичных продуктов соответствующих генов генные болезни подразделяют на наследственные нарушения ферментных систем (энзимопатии), дефекты белков крови (гемоглобинопатии), дефекты структурных белков (коллагеновые болезни) и генные болезни с невыясненным первичным биохимическим дефектом. Энзимопатии. В основе энзимопатии лежат либо изменения активности фермента, либо снижение интенсивности его синтеза. У гетерозигот-носителей мутантного гена присутствие нормального аллеля обеспечивает сохранение около 50% активности фермента по сравнению с нормальным состоянием. Поэтому наследственные дефекты ферментов клинически проявляются у гомозигот, а у гетерозигот недостаточная активность фермента выявляется специальными исследованиями. В зависимости от характера нарушения обмена веществ в клетках среди энзимопатий различают следующие формы. 1. Наследственные дефекты обмена углеводов (галактоземия — нарушение метаболизма молочного сахара — лактозы; мукополисаха-ридозы — нарушение расщепления полисахаридов). 2. Наследственные дефекты обмена липидов и липопротеинов (сфинголипидозы — нарушение расщепления структурных липидов; нарушения обмена липидов плазмы крови, сопровождающиеся увеличением или снижением в крови холестерина, лецитина). 3. Наследственные дефекты обмена аминокислот (фенилкетонурия —нарушение обмена фенилаланина (см. разд. 4.1); тирозиноз— нарушение обмена тирозина; альбинизм — нарушение синтеза пигмента меланина из тирозина и др.). 4. Наследственные дефекты обмена витаминов (гомоцистинурия — развивается как результат генетического, дефекта кофермента витаминов В6 и B12, наследуется по аутосомно-рецессивному типу). 5. Наследственные дефекты обмена пуриновых и пиримидиновых азотистых оснований (синдром Леша — Найяна, связанный с недостаточностью фермента, который катализирует превращение свободных пуриновых оснований в нуклеотиды, наследуется по Х-сцепленному рецессивному типу). 6. Наследственные дефекты биосинтеза гормонов (адреногенитальный синдром, связанный с мутациями генов, которые контролируют синтез андрогенов; тестикулярная феминизация, при которой не образуются рецепторы андрогенов). 7. Наследственные дефекты ферментов эритроцитов (некоторые гемолитические несфероцитарные анемии, характеризующиеся нормальной структурой гемоглобина, но нарушением ферментной системы, участвующей в анаэробном (бескислородном) расщеплении глюкозы. Наследуются как по аутосомно-рецессивному, так и по Х-сцепленному рецессивному типу). Гемоглобинопатии. Это группа наследственных заболеваний, вызываемых первичным дефектом пептидных цепей гемоглобина и связанным с этим нарушением его свойств и функций. К ним относят метгемоглобинемии, эритроцитозы, серповидно-клеточную анемию, талассемии (см. § 4.1). Коллагеновые болезни. В основе возникновения этих заболеваний лежат генетические дефекты биосинтеза и распада коллагена — важнейшего структурного компонента соединительной ткани. К этой группе относят болезнь Эллерса — Данлоса, характеризующуюся большим генетическим полиморфизмом и наследующуюся как по аутосомно-доминантному, так и по аутосомно-рецессивному типу, болезнь Марфана, наследующуюся по аутосомно-доминантному типу, и ряд других заболеваний. Наследственные болезни с невыясненным первичным биохимическим дефектом. К этой группе принадлежит подавляющее большинство моногенных наследственных болезней. Наиболее распространенными являются следующие. 1. Муковисцидозы — встречаются с частотой 1:2500 новорожденных; наследуются по аутосомно-рецессивному типу. В основе патогенеза заболевания —наследственное поражение экзокринных желез и железистых клеток организма, выделение ими густого, измененного по составу секрета и связанные с этим последствия. 2. Ахондроплазия — заболевание, в 80—95% случаев обусловленное вновь возникшей мутацией; наследуется по аутосомно-доминантному типу; встречается с частотой приблизительно 1:100 000. Это заболевание костной системы, при котором наблюдаются аномалии развития хрящевой ткани преимущественно в эпифизах трубчатых костей и костях основания черепа (рис. 6.23). 3. Мышечные дистрофии (миопатии) —заболевания, связанные с поражением поперечно-полосатых и гладких мышц. Различные формы характеризуются разным типом наследования. Например, прогрессирующая псевдогипертрофическая миопатия Дюшена наследуется по Х-сцепленному рецессивному типу и проявляется преимущественно у мальчиков в начале первого десятилетия жизни. Известна мышечная псевдогипертрофическая дистрофия, наследующаяся по аутосомно-рецессивному типу, которая начинает развиваться во второй половине первого десятилетия жизни и встречается с одинаковой частотой у обоих полов. Мышечная дистрофия плечевого и тазового пояса: наследуется по аутосомно-доминантному типу и т.д. Генетическое многообразие генных болезней. Изучение наследственных заболеваний у человека свидетельствует о том, что нередко сходное фенотипическое проявление болезни бывает обусловлено несколькими различными мутациями. Это явление впервые было описано в 30-х гг. С. Н. Давиденковым и названо генетической гетерогенностью наследственных заболеваний. Генетическая гетерогенность наследственных болезней может быть обусловлена мутациями разных генов, кодирующих ферменты одного метаболического пути, а также мутациями одного и того же гена, приводящими к появлению разных его аллелей. Среди рассмотренных выше наследственных болезней особенно высокой степенью генетического полиморфизма отличаются мукопо-лисахаридозы, генетическая разнородность которых объясняется множественными мутациями в 11—12 генах, связанных общей функцией расщепления полисахаридов. Большой генетической гетерогенностью характеризуется врожденная аутосомно-рецессивная форма глухоты, при которой различают не менее 35 генетически различных вариантов с фенотипически сходным проявлением. Большие перспективы в расшифровке наследственной гетерогенности генных болезней открываются в связи с применением молекулярно-генетических методов их прямого анализа с помощью ДНК-зондов. Есть моногенные и полигенные болезни. Моногенные болезни наследственного предрасположения – наследственные заболевания, проявляющиеся из-за мутации одного гена или проявляющиеся при действии определенного фактора среды (аутосомно-рецессивные или сцепленные с Х-хромосомой). Проявляются при воздействии факторов: - физических; - химических; - пищевых; - загрязнения среды. Парамиотомия – в сырую погоду происходят тонические спазмы мышц при холоде, под влиянием тепла – проходят. Болезнь связана с термочувствительным белком. Реакция проявляется в младенчестве и не изменяется на протяжении жизни человека. Пигментная ксеродерма - веснушчатая кожа особого типа. Проявляется в 4-6 лет. Дети не переносят УФ-свет возникают злокачественные опухоли, такие дети умирают от метастаз еще до 15 лет. Не переносят также и гамма-лучей. Синдром Блюма. Пигментная «бабочка» на лице, маленький рост, удлиненная голова. Евреи, поляки, беларусы, австрийцы. Погибают до 18 лет. Не переносят УФ-облучения, гамма-лучей. Альфа-1 антитрипсин при загрязнении воздуха, табачном дыме проявляется острой закупоркой бронхов или циррозом печени. У европеоидов люди, не переносящие молоко, составляют 10-20%, в Африке – 70-80%. Влияние лекарственных средств: сульфаниламидные препараты провоцируют заболевания крови. Есть полигенные болезни наследственного происхождения – такие болезни, которые возникают при действии многих факторов (мультифакториальные) и в результате взаимодействия многих генов. Установить диагноз в таком случае очень сложно, т.к. действует много факторов, и появляется новое качество при взаимодействии факторов. 47.Хромосомные болезни человека, механизмы их возникновения и проявления. Примеры. Хромосомные болезни, наследственные заболевания, обусловленные изменением числа или структуры хромосом. Эта группа заболеваний обусловлена изменением структуры отдельных хромосом или их количества в кариотипе. Как правило, при таких мутациях наблюдается дисбаланс наследственного материала, который и ведет к нарушению развития организма. У человека описаны геномные мутации по типу полиплоидии, которые редко наблюдаются у живорожденных, а в основном обнаруживаются у абортированных эмбрионов и плодов и у мертворожденных. Основную часть хромосомных болезней составляют анэуплоидии, причем моносомии по аутосомам у живорожденных встречаются крайне редко. Большинство из них касаются 21-й и 22-й хромосом и чаще обнаруживаются у мозаиков, имеющих одновременно клетки с нормальным и мутантным кариотипом. Достаточно редко обнаруживается моносомия и по Х-хромосоме (синдром Шерешевского — Тернера). В отличие от моносомии трисомии описаны по большому числу аутосом: 8, 9, 13, 14, 18, 21, 22-й и Х-хромосоме, которая может присутствовать в кариотипе в 4—5 экземплярах, что вполне совместимо с жизнью. Структурные перестройки хромосом также, как правило, сопровождаются дисбалансом генетического материала (делеции, дупликации). Степень снижения жизнеспособности при хромосомных аберрациях зависит от количества недостающего или избыточного наследственного материала и от вида измененной хромосомы. К настоящему времени описано около 100 клинико-цитогенетических синдромов, в основе которых лежат различные хромосомные аномалии. Хромосомные изменения, приводящие к порокам развития, чаще всего привносятся в зиготу с гаметой одного из родителей при оплодотворении. При этом все клетки нового организма будут содержать аномальный хромосомный набор и для диагностики такого заболевания достаточно проанализировать кариотип клеток какой-нибудь ткани. Если хромосомные нарушения возникают в одном из бластомеров во время первых делений зиготы, образующейся из нормальных гамет, то развивается мозаичный организм, большая или меньшая часть клеток которого несет нормальный хромосомный набор. Диагностика мозаичных форм хромосомных болезней отличается большей трудоемкостью и требует изучения кариотипа большого числа клеток из разных тканей. Для определения вероятности появления хромосомной болезни в потомстве в семьях, уже имеющих больных детей, важно установить, является ли это хромосомное нарушение заново возникшим или оно унаследовано от предыдущего поколения. Чаще родители человека с хромосомным заболеванием имеют нормальный кариотип, а появление больного потомства является результатом мутации, возникшей в одной из гамет. В этом случае возможность повторного хромосомного нарушения у детей в данной семье маловероятна и не превосходит таковой в целом для популяции. Вместе с тем описано немало семей, в которых наблюдается предрасположение, например, к нерасхождению хромосом. В случае наследуемых хромосомных болезней в соматических клетках родителей обнаруживаются хромосомные или геномные мутации, которые могут передаваться их зрелым половым клеткам в ходе гаметогенеза. Передают потомству хромосомные нарушения обычно фенотипически нормальные родители, являющиеся носителями сбалансированных хромосомных перестроек — реципрокных транслокаций, робертсоновских транслокаций или перицентрических инверсий. У носителей такого рода хромосомных перестроек с определенной вероятностью образуются нормальные гаметы, а также гаметы, несущие сбалансированную перестройку, и половые клетки с нарушенным балансом генов в геноме (рис. 6.22). 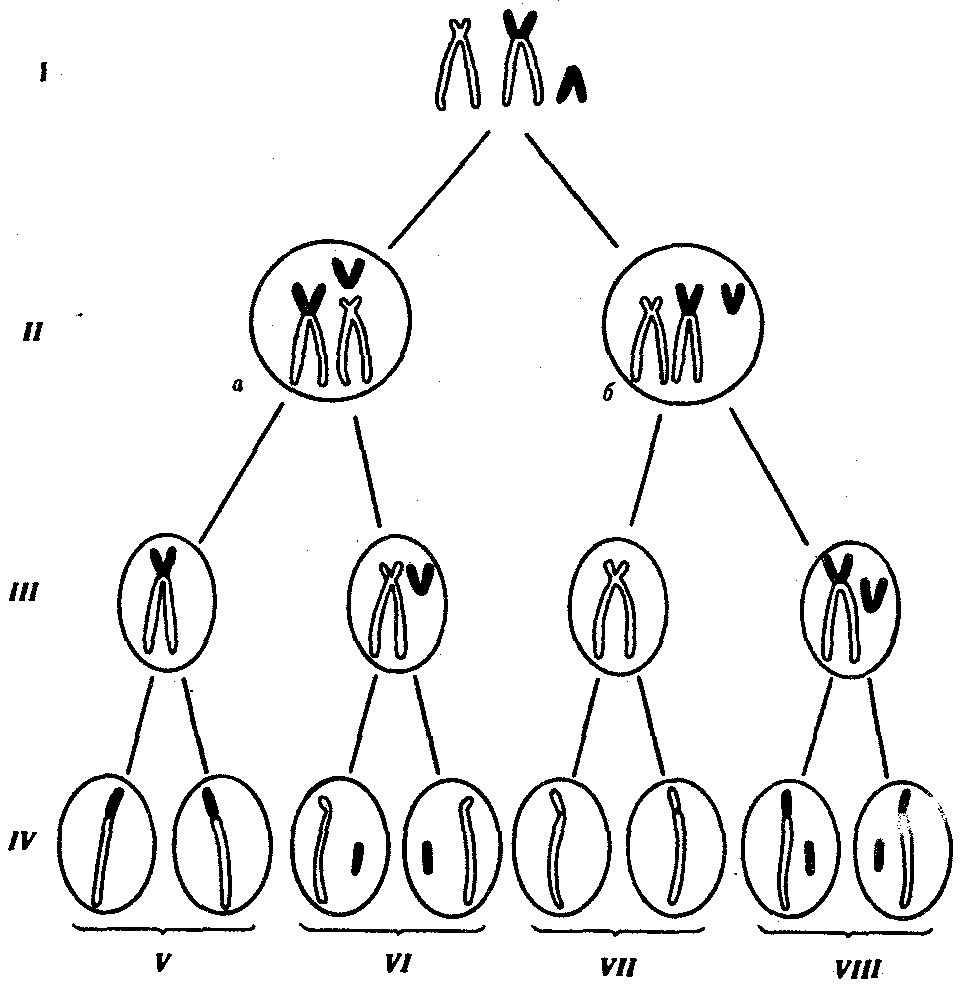 Рис. 6.22. Вероятность образования нормальных и аномальных гамет у носителей сбалансированной хромосомной перестройки Показана робертсоновская транслокация 21-й хромосомы (окрашена) на одну из акродентрических хромосом (не окрашена); I — кариотип носителя сбалансированной хромосомной перестройки, II — варианты (а, б) расположения бивалентов в экваториальной плоскости веретена деления (метафаза I), III — результат 1-го редукционного деления мейоза; IV — результат 2-го эквационного деления мейоза; V — гаметы со сбалансированной хромосомной перестройкой; VI — нормальные гаметы; VII — гаметы, не имеющие 21-й хромосомы; VIII — гаметы, содержащие две 21-е хромосомы (VII и VIII — гаметы с несбалансированным геномом) Возможность наследования хромосомных аномалий делает необходимым анализ кариотипа родителей, уже имеющих больных детей, и пренатальную диагностику развивающегося внутриутробно плода для исключения вероятности повторного рождения ребенка с хромосомной болезнью. Фенотипическое проявление различных хромосомных и геномных мутаций характеризуется ранним и множественным поражением различных систем органов. Типичными являются задержка общего физического и умственного развития, отклонения в строении скелета, в частности мозгового и лицевого черепа, пороки развития сердечно-сосудистой, мочеполовой, нервной систем, нарушения в биохимическом, гормональном и иммунологическом статусе организма. Хромосомные болезни, как правило, характеризуются сочетанием многих врожденных пороков. Для них также характерны многообразие и вариабельность фенотипических проявлений. Наиболее специфические проявления хромосомных заболеваний связаны с дисбалансом по относительно небольшому фрагменту хромосомы. Так, фенотипическое проявление синдрома Дауна наблюдается в случае трисомии всего лишь по небольшому сегменту длинного плеча 21-й хромосомы. Картина синдрома «кошачьего крика» развивается при утрате участка короткого плеча 5-й хромосомы. Дисбаланс по значительному объему хромосомного материала делает фенотипическую картину менее специфической. Специфичность проявления хромосомного заболевания определяется изменением содержания определенных структурных генов, кодирующих синтез специфических белков. Так, при болезни Дауна обнаружено повышение в 1,5 раза активности фермента супероксид-дисмутазы I, ген которого располагается в 21-й хромосоме и представлен у больных в трехкратной дозе. Эффект «дозы гена» обнаружен более чем для 30 генов, локализованных в разных хромосомах человека. Полуспецифические симптомы проявления хромосомных болезней связаны в значительной мере с дисбалансом генов, представленных многими копиями, которые контролируют ключевые процессы в жизнедеятельности клеток и кодируют, к примеру, структуру рРНК, тРНК, гистонов, рибосомальных белков, актина, тубулина. Неспецифические проявления при хромосомных болезнях связывают с изменением содержания гетерохроматина в клетках, который оказывает влияние на нормальное течение клеточного деления и роста, формирование в онтогенезе количественных признаков, определяемых полигенами. Для медицинской практики в 1971 году был проведен симпозиум по медицинской генетике в Париже. Была принята международная Парижская классификация для обозначения кариотипа человека. 46,хх; 46,ху – кариотип нормального человека. Во время мейоза возможно появление аномальных половых клеток. 47,хху – синдром Клайнфельтера. Мужчина, частота встречаемости 1 из 1000 новорожденных мальчиков. Высокий рост, более длинные ноги, евнуховидное телосложение, недоразвитие половых органов, гинекомастия, у половины умственная отсталость (трудности в обучении чтению и письму), могут заканчивать нормальные школы, хотя им может быть очень трудно. Вспыльчивы, импульсивны, легко попадают од влияние более сильных личностей, преступления и проступки. Жизнеспособность снижена. Среди «туповатых» преступников приблизительно 2%. 47,хуу – синдром двойного игрек (трисомия) 1 на 700 новорожденных. Впервые в 1977году были исследованы. Высокие мужчины, агрессивное поведение, интеллект снижен или находится на нижней границе нормы. Характерные преступления – поджоги, воровство, детоубийство без мотивации. В больницах закрытого типа, в колонии – 5% таких людей. Поведение детерминировано лишней хромосомой. 47,ххх – синдром Сверхженщины. 1на 1000 новорожденных девочек. Внешне не проявляется, легкое слабоумие. Считают, что около 1% девушек и женщин с легким слабоумием. Могут беременеть и рождают нормальных детей (во время мейоза происходит самокоррекция). 45,у0 – нежизнеспособны – аборт. 45,х0 синдром Шеришевкого-Тернера частота встречаемости 1:2000 девочек. Летальность при моносомии очень высокая, каждый 13 выкидыш имеет такую природу. Фенотипические проявления – маленький рост, для многих характерна шейная складка. Локтевой изгиб под углом, укорочены 4 и 5 пальцы, антимонголоидные глаза, абстрактное мышление отсутствует, упорные, трудолюбивые, способны заканчивать школы, ВУЗы. Любовь к опеканию маленьких детей. Отсутствует критическое восприятие своих дефектов. Низкий рост девочки – непременное условие для проведения кариотипирования. Окружность головы больше нормы, груди широко расставлены. 49,ххххх – нарушения те же, Но встречаемость ниже 49,хххху – то же. Аутосом меньше 44 не бывает, но больше – возможно. 47,хх+21, 47,ху+21 Синдром Дауна. Частота встречаемости 1на 650 новорожденных. Фенотипических признаков очень много. Большой язык. Не помещающийся в полости рта, специфический разрез глаз, умственная отсталость и т.д. 12% умственно отсталых детей - Дауны. Частота встречаемости у девочек и мальчиков разных рас примерно одинакова. Чем старше мать, тем выше вероятность рождения ребенка с этой патологией. Каждый 40 ребенок после 40 лет. Не способны к трудовой деятельности, требуют ухода и дорогостоящего лечения. 47,хх+13,47,ху+13 Синдром Патау. 1 больной на 7-8 тысяч новорожденных. Новорожденные имеют нормальные вес и рост. Характерны микроцефалия (недоразвитие головного мозга), резкая умственная отсталость, незарощение неба и губы. Полидактилия, повышенная гибкость суставов, недоразвитие глазного яблока, неправильно сформированные, низко посаженные ушные раковины, пороки внутренних органов. Такие дети не живут долго. 47,хх+18, 47,ху+18 Синдром Эдвардса. Частота встречаемости у девочек в 3 раза выше, чем у мальчиков. 1 больной на 6-7 тысяч новорожденных. Характерны множественные аномалии, грубые пороки, характерна грубая задержка роста (гипоплазия в эмбриональном периоде), своеобразный свод черепа, пяткообразно нависающий затылок, короткая шея, расстояние между висками маленькое, ушная раковина деформирована, у половины на затылке избыточная кожа. Продолжительность жизни таких детей снижена. 10% погибают до 1 месяца, 19=0% - до 3 и 30% погибают до года. Трисомии могут быть по любой хромосоме. Большей частью по 1 паре аутосом. Чем больше генетического материала, тем хуже. В первую очередь страдает интеллект. Клеточный мозаицизм (генетический) – в соматических клетках одного и того же организма имеется разный набор хромосом. Возникает в результате нерасхождения хромосом во время митоза. По наследству не передается. Проявление зависит от соотношения клеток. Структурные аномалии хромосом. Изохромосомы – разделение хромосомы неправильным путем. Чем больше возраст отца, тем, чаще встречается подобное нарушение. 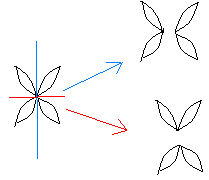 46,isoX Дилеция (частичная моносомия) Р – длинное плечо, Q – короткое. 46,хх,5р – дилеция плеча5 хромосомы. Синдром Кошачий крик. Широко расставленные глаза, физическое недоразвитие. Множественные пороки развития, недоразвита гортань – специфический крик. Транслокация – обмен участками хромосом (3 вида). Реципроксные (обмен участками между негомологичными хромосомами). 46,ху,t(9,22) – миелолейкоз (рак крови). Нереципроксные (между 2мя гомологичными хромосомами). Может не проявляться. Робертсоновские: возникают при нарушениях деления акроцентрических хромосом. Разрыв по центромере, короткие части дегенерируют, длинные срастаются часто по 15 хромосоме. 46,хх,15t – рак крови. Приводит к ожирению, гипотонии мышц, умственной отсталости. Возможно рождение ребенка – Дауна(5-10% перенос с 21 на 14). Инверсия – поворот. Кольцевые хромосомы могут возникать по 16и 18 хромосомам, терминальные концы разрываются. Обозначается – Г. По 18 хромосоме – слабоумие, аномалии лица. В результате хромосомных мутаций и аббераций возникает дисбаланс генетического материала, что приводит к психическим и физическим нарушениям развития. Аномалии по крупным хромосомам происходят значительно реже, чем по мелким. Самая маленькая хромосома – 21, нарушения ее строения встречаются чаще всего. Нехватка генетического материала переносится хуже, чем избыток. Если много эухроматина – нежизнеспособность ребенка, если преобладает гетерохроматин – тяжелые патологии (8,13,18,21,х хромосомы). |