Патологическая анатомия содержание, задачи, объекты и методы исследования. Патологическая анатомия
 Скачать 1.54 Mb. Скачать 1.54 Mb.
|

|
27) Аутоиммунизация: определение. Аутоиммунные болезни, классификация, механизмы развития, морфологическая характеристика. Аутоиммунизация (аутоаллергия, аутоагрессия) - состояние, характеризующееся появлением реакции иммунной системы на нормальные антигены собственных тканей. Аутоиммунизация тесно связана с понятием иммунологической толерантности (от лат. tolerare - переносить, терпеть). Оно характеризуется состоянием ареактивности («терпимости») лимфоидной ткани по отношению к антигенам, способным вызывать иммунный ответ. В период созревания лимфоидной ткани возникает иммунологическая толерантность к антигенам всех органов и тканей, кроме тканей глаза, щитовидной железы, семенников, надпочечников, головного мозга и нервов. Считается, что антигены этих органов и тканей отграничены от лимфоидной ткани физиологическими барьерами, что и объясняет отсутствие к ним толерантности иммунокомпетентной системы. «Свои» и «чужие» тканевые антигены иммунная система начинает распознавать у новорожденного через несколько недель после рождения. Среди этиологических факторов аутоиммунизации ведущее значение придается хронической вирусной инфекции, радиации и генетическим нарушениям. Этиология тесно связана с патогенезом. В патогенезеаутоиммунных заболеваний различают предрасполагающие, инициирующие и способствующие факторы. Кпредрасполагающим факторам относят определенные гены системы HLA, определяющие количественные и качественные индивидуальные особенности иммунного ответа; гормональный фон, связанный прежде всего с полом (у женщин аутоиммунные болезни встречаются в 6-9 раз чаще, чем у мужчин), и генетически обусловленные особенности клеток органов-мишеней аутоиммунного процесса. Неблагоприятное сочетание этих факторов определяет 50% риска развития болезни. Инициирующими факторами могут быть вирусные и бактериальные инфекции, физические, химические воздействия как на органы иммунной системы, так и на органы-мишени. Способствующие факторы аутоиммунизации - это дисфункция иммунной системы - снижение супрессорной активности Т-лимфоцитов и антиидиотипических антител. Аутоиммунные болезни - это болезни, в основе которых лежит аутоиммунизация, т.е. агрессия аутоантител, циркулирующих иммунных комплексов, содержащих аутоантигены, и эффекторных иммунных клеток (лимфоцитов-киллеров) в отношении антигенов собственных тканей организма. Поэтому аутоиммунные болезни называют также аутоагрессивными. Руководствуясь механизмом аутоиммунизации, различают две группы аутоиммунных заболеваний. Первая группа - это органоспецифические аутоиммунные болезни, которые развиваются в связи с повреждением физиологических барьеров иммунологически обособленных органов, что позволяет иммунной системе реагировать на их неизмененные антигены выработкой аутоантител и сенсибилизированных лимфоцитов. При этом в органах развиваются морфологические изменения, характерные преимущественно для ГЗТ: ткань органов инфильтрируется лимфоцитами, паренхиматозные элементы погибают, в финале развивается склероз. В эту группу входят тиреоидит (болезнь Хасимото) (рис. 80), энцефаломиелит, полиневрит, рассеянный склероз, идиопатическая аддисонова болезнь, асперматогения, симпатическая офтальмия. Вторая группа - это органонеспецифические аутоиммунные заболевания. Ведущими при этих заболеваниях являются нарушения контроля иммунологического гомеостаза лимфоидной системой. Аутоиммунизация при этом развивается по отношению к антигенам многих органов и тканей, не обладающих органной специфичностью и неспособных вызывать продукцию антител при парентеральном введении. В органах и тканях развиваются морфологические изменения, характерные для реак ций гиперчувствительности как замедленного, так и особенно немедленного типов. К этой группе аутоиммунных заболеваний относят системную красную волчанку, ревматоидный артрит, системную склеродермию, дерматомиозит (группа ревматических болезней), вторичную тромботическую тромбоцитопеническую пурпуру (болезнь Мошковича). Существуют аутоиммунные болезни промежуточного типа, т.е. близкие аутоиммунным заболеваниям первого или второго типа. Это миастения гравис, сахарный диабет I типа, тиреотоксикоз, синдромы Шегрена и Гудпасчера и др. Помимо аутоиммунных заболеваний, выделяют болезни с аутоиммунными нарушениями. Появление аутоантигенов при этих заболеваниях связывают с изменением антигенных свойств тканей и органов - денатурацией тканевых белков (при ожоге, облучении, травме, хроническом воспалении, вирусной инфекции); образование аутоантигена возможно при воздействии бактериального антигена, особенно перекрестно реагирующего (например, при гломерулонефрите, ревматизме). В образовании аутоантигена большое значение придается гаптенному механизму, причем в роли гаптена могут выступать как продукты обмена тела, так и микроорганизмы, токсины и лекарственные средства. Аутоиммунизация в этих условиях определяет не возникновение заболевания, а прогрессирование характерных для него локальных (органных) изменений, которые отражают «морфологию реакций гиперчувствительности замедленного и немедленного типов. В эту группу заболеваний включают: определенные формы гломерулонефрита, гепатита, хронического гастрита и энтерита, цирроз печени, ожоговую болезнь, аллергические анемии, тромбоцитопению, агранулоцитоз, лекарственную аллергию. 28) Иммунодефицитные синдромы, их клинико-морфологические проявления. Иммунодефицитные синдромы являются крайним проявлением недостаточности иммунной системы. Они могут быть первичными, обусловленными недоразвитием (гипоплазия, аплазия) иммунной системы -наследственные и врожденные иммунодефицитные синдромы, или вторичными (приобретенными), возникающими в связи с болезнью или проводимым лечением. Первичные иммунодефицитные синдромы Первичные иммунодефицитные синдромы могут быть выражением недостаточности: 1) клеточного и гуморального иммунитета; 2) клеточного иммунитета; 3) гуморального иммунитета. Синдромы недостаточности клеточного и гуморального иммунитета называют комбинированными. Они встречаются у детей и новорожденных, наследуются по аутосомно-доминантному типу (агаммаглобулинемия швейцарского типа, или синдром Гланцманна-Риникера; атаксиятелеангиэктазия Луи-Бар). При этих синдромах находят гипоплазию как вилочковои железы, так и периферической лимфоидной ткани (табл. 4), что и определяет дефект клеточного и гуморального иммунитета. В связи с несостоятельностью иммунитета у таких детей часто возникают инфекционные заболевания, которые имеют рецидивирующее течение и дают тяжелые осложнения (пневмония, менингит, сепсис), отмечается задержка физиологического развития. При комбинированных иммунодефицитных синдромах часто возникают пороки развития и злокачественные мезенхимальные опухоли (атаксия-телеангиэктазия Луи-Бар). Синдромы недостаточности клеточного иммунитета в одних случаях наследуются обычно по аутосомно-доминантному типу (иммунодефицит с ахондроплазией, или синдром Мак-Кьюсика), в других являются врожденными (агенезия или гипоплазия вилочковои железы, или синдром Дайджорджа). Помимо агенезии или гипоплазии вилочковои железы и Т-зависимых зон периферической лимфоидной ткани, что определяет дефицит клеточного иммунитета, для этих синдромов характерны множественные пороки развития (см. табл. 4). Дети погибают от пороков развития либо от осложнений инфекционных заболеваний. Синдромы недостаточности гуморального иммунитета имеют наследственную природу, причем установлена сцепленность их с Х-хромосомой (см. табл. 4). Болеют дети первых пяти лет жизни. Для одних синдромов (агаммаглобулинемия, сцепленная с Х-хромосомой, или синдром Брутона) характерна потеря способности к синтезу всех иммуноглобулинов, что морфологически подтверждается отсутствием В-зависимых зон и клеток плазмоцитарного ряда в периферической лимфоидной ткани, прежде всего в лимфатических узлах и селезенке. Другим синдромам свойствен дефицит одного из иммуноглобулинов (например, избирательный дефицит IgA, или синдром Веста), тогда структура лимфоидной ткани остается сохранной. Однако при всех синдромах недостаточности гуморального иммунитета развиваются тяжелые бактериальные инфекции с преобладанием гнойно-деструктивных процессов в бронхах и легких, желудочнокишечном тракте, коже, ЦНС, нередко заканчивающихся сепсисом. Помимо иммунодефицитных, известны синдромы недостаточности системы моноритарных фагоцитов и нейтрофилов, среди которых наследственные заболевания и синдромы - хроническая гранулематозная болезнь, синдромы Чедиака-Хигаси и Джоба и др. Вторичные иммунодефицитные синдромы Вторичные (приобретенные) иммунодефицитные синдромы в отличие от первичных возникают в связи с болезнью или определенным видом лечения. Среди заболеваний, ведущих к развитию недостаточности иммунной системы, основное значение имеет безудержно распространяющийся во многих странах мира синдром приобретенного иммунного дефицита, или СПИД, - самостоятельное заболевание, вызываемое определенным вирусом (см. Вирусные инфекции). К развитию вторичных иммунодефицитных синдромов ведут также другие инфекции, лейкозы, злокачественные 208 лимфомы (лимфогранулематоз, лимфосаркома, ретикулосаркома), тимома, саркоидоз. При этих заболеваниях возникает недостаточность гуморального и клеточного иммунитета в результате дефекта популяции как В-, так и Т-лимфоцитов, а возможно, и их предшественников. Среди видов лечения, ведущих к вторичной недостаточности иммунной системы, наибольшее значение имеют лучевая терапия, применение кортикостероидов и иммунодепрессантов, антилимфоцитарной сыворотки, тимэктомия, дренирование грудного протока и др. Недостаточность иммунной системы, развивающаяся в связи с лечением той или иной болезни, рассматривается как патология терапии (ятрогения). При вторичных, как и при первичных, иммунодефицитных синдромах часто наблюдаются гнойные инфекции, обострение туберкулезного процесса, сепсис. 29) Амилоидоз: определение, классификация, морфологическая характеристика. Амилоидоз (от лат, amylum - крахмал), или амилоидная дистрофия, - стромально-сосудистый диспротеиноз, сопровождающийся глубоким нарушением белкового обмена, появлением аномального фибриллярного белка и образованием в межуточной ткани и стенках сосудов сложного вещества - амилоида. Классификация амилоидоза учитывает следующие признаки: 1) возможную причину; 2) специфику белка фибрилл амилоида; 3) распространенность амилоидоза; 4) своеобразие клинических проявлений в связи с преимущественным поражением определенных органов и систем. 1. Руководствуясь причиной, выделяют -первичный (идиопатический), -наследственный (генетический, семейный), -вторичный (приобретенный) -старческий амилоидоз. Первичный, наследственный, старческий амилоидозы рассматривают в качестве нозологических форм. Вторичный амилоидоз, встречающийся при тех или иных заболеваниях, является осложнением этих заболеваний, «второй болезнью». 2. Специфика белка фибрилл амилоида позволяет выделить AL-, АА-, AF- и ASC1-амилоидоз. 3. Учитывая распространенность амилоидоза, различают генерализованную и локальную формы. К генерализованному амилоидозу относят первичный амилоидоз и амилоидоз при «плазмоклеточной дискразии» (формы AL-амилоидоза), вторичный амилоидоз и некоторые типы наследственного (формы АА-амилоидоза), а также старческий системный амилоидоз (ASC1-амилоидоз). Локальный амилоидоз объединяет ряд форм наследственного и старческого амилоидоза, а также локальный опухолевидный амилоидоз («амилоидная опухоль»). 4. Своеобразие клинических проявлений в связи с преимущественным поражением органов и систем позволит выделять кардиопатический, нефропатический, нейропатический, гепатопатический, эпинефропатический, смешанный типы амилоидоза и APUD-амилоидоз. Кардиопатический тип чаще встречается при первичном и старческом системном амилоидозе, нефропатический - при вторичном амилоидозе, периодической болезни и болезни Маккла и Уэллса; для вторичного амилоидоза характерны и смешанные типы (сочетание поражения почек, печени, надпочечников, желудочно-кишечного тракта). Нейропатический амилоидоз, как правило, имеет наследственный характер. APUD-амилоид развивается в органах APUD-системы при развитии в них опухолей (апудом), а также в островках поджелудочной железы при старческом амилоидозе. Морфо- и патогенез амилоидоза. Функцию амилоидобластов, продуцирующих белок фибрилл амилоида (рис. 34), при различных формах амилоидоза выполняют разные клетки. При генерализованных формах амилоидоза - это главным образом макрофаги, плазматические и миеломные клетки; однако не исключается роль фибробластов, ретикулярных клеток и эндотелиоцитов. При локальных формах в роли амилоидобластов могут выступать кардиомиоциты (амилоидоз сердца), гладкие мышечные клетки (амилоидоз аорты), кератиноциты (амилоидоз кожи), В-клетки островков поджелудочной железы (инсулярный амилоидоз), С-клетки щитовидной железы и другие эпителиальные клетки APUD-системы. Амилоидные отложения имеют довольно типичную локализацию: в стенках кровеносных и лимфатических капилляров и сосудов в интиме или адвентиции; в строме органов по ходу ретикулярных и коллагеновых волокон; в собственной оболочке железистых структур. Амилоидные массы вытесняют и замещают паренхиматозные элементы органов, что ведет к развитию их хронической функциональной недостаточности. Патогенез амилоидоза сложен и неоднозначен у различных его форм и типов. Лучше других форм изучен патогенез АА- и AL-амилоидоза. При АА-амилоидозе фибриллы амилоида образуются из поступающего в макрофаг - амилоидобласт плазменного предшественника фибриллярного белка амилоида - белка SAA, который усиленно синтезируется в печени (схема III). Усиленный синтез SAA гепатоцитами стимулирует макрофагальный медиаторинтерлейкин-1, что приводит к резкому увеличению содержания SAA в крови (предамилоидная стадия). В этих условиях макрофаги не в состоянии осуществить полную деградацию SAA, и из фрагментов в инвагинатах плазматической мембраны амилоидобласта происходит сборка фибрилл амилоида (см. рис. 34). Стимулирует эту сборку амилоидстимулирующий фактор (АСФ), который обнаруживается в тканях (селезенка, печень) в предамилоидной стадии. Таким образом, ведущую роль в патогенезе АА-амилоидоза играет макрофагальная система: она стимулирует усиленный синтез белка предшественника - SAA печенью, она же участвует и в образовании фибрилл амилоида из деградирующих фрагментов этого белка. При AL-амилоидозе сывороточным предшественником белка амилоидных фибрилл являются L-цепи иммуноглобулинов. Считают, что возможны два механизма образования AL-амилоидных фибрилл: 1) нарушение деградации моноклоновых легких цепей с образованием фрагментов, способных к агрегации в амилоидные фибриллы; 2) появление L-цепей с особыми вторичными и третичными структурами при аминокислотных заменах. Синтез амилоидных фибрилл из L-цепей иммуноглобулинов может происходить не только в макрофагах, но и в плазматических и миеломных клетках, синтезирующих парапротеины (схема IV). Таким образом, к патогенезу AL-амилоидоза причастна прежде всего лимфоидная система; с ее извращенной функцией связано появление «амилоидогенных» легких цепей иммуноглобулинов - предшественника амилоидных фибрилл. Роль макрофагальной системы при этом вторичная, соподчиненная. Макро- и микроскопическая характеристика амилоидоза. Внешний вид органов при амилоидозе зависит от степени процесса. Если отложения амилоида небольшие, внешний вид органа изменяется мало и амилоидоз обнаруживается лишь при микроскопическом исследовании. При выраженном амилоидозе орган увеличивается в объеме, становится очень плотным и ломким, а на разрезе имеет своеобразный восковидный, или сальный, вид. 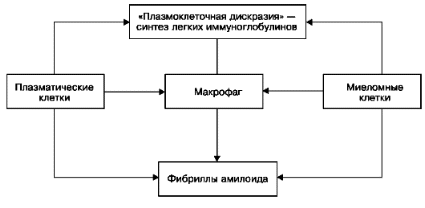 В селезенке амилоид откладывается в лимфатических фолликулах (рис. 35) или же равномерно по всей пульпе. В первом случае амилоидноизмененные фолликулы увеличенной и плотной селезенки на разрезе имеют вид полупрозрачных зерен, напоминающих зерна саго (саговая селезенка). Во втором случае селезенка увеличена, плотная, коричнево-красная, гладкая, имеет сальный блеск на разрезе (сальная селезенка). Саговая и сальная селезенка представляют последовательные стадии процесса. В почках амилоид откладывается в стенке сосудов, в капиллярных петлях и мезангии клубочков, в базальных мембранах канальцев и в строме. Почки становятся плотными, большими и «сальными». По мере нарастания процесса клубочки и пирамиды полностью замещаются амилоидом (см. рис. 35), разрастается соединительная ткань и развивается амилоидное сморщивание почек. В печени отложение амилоида наблюдается между звездчатыми ретикулоэндотелиоцитами синусоидов, по ходу ретикулярной стромы долек, в стенках сосудов, протоков и в соединительной ткани портальных трактов. По мере накопления амилоида печеночные клетки атрофируются и погибают. При этом печень увеличена, плотная, выглядит «сальной». В кишечнике амилоид выпадает по ходу ретикулярной стромы слизистой оболочки, а также в стенках сосудов как слизистой оболочки, так и подслизистого слоя. При резко выраженном амилоидозе железистый аппарат кишечника атрофируется. Амилоидоз надпочечников, как правило, двусторонний, отложение амилоида встречается в корковом веществе по ходу сосудов и капилляров. В сердце амилоид обнаруживается под эндокардом, в строме и сосудах миокарда (см. рис. 35), а также в эпикарде по ходу вен. Отложение амилоида в сердце ведет к резкому его увеличению (амилоидная кардиомегалия). Оно становится очень плотным, миокард приобретает сальный вид. В скелетных мышцах, как и в миокарде, амилоид выпадает по ходу межмышечной соединительной ткани, в стенках сосудов и в нервах. Периваскулярно и периневрально нередко образуются массивные отложения амилоидного вещества. Мышцы становятся плотными, полупрозрачными. 85 В легких отложения амилоида появляются сначала в стенках разветвлений легочных артерии и вены (см. рис. 35), а также в перибронхиальной соединительной ткани. Позже амилоид появляется в межальвеолярных перегородках. В головном мозге при старческом амилоидозе амилоид находят в сенильных бляшках коры, сосудах и оболочках. Амилоидоз кожи характеризуется диффузным отложением амилоида в сосочках кожи и ее ретикулярном слое, в стенках сосудов и по периферии сальных и потовых желез, что сопровождается деструкцией эластических волокон и резкой атрофией эпидермиса. Амилоидоз поджелудочной железы имеет некоторое своеобразие. Помимо артерий железы, встречается и амилоидоз островков, что наблюдается в глубокой старости. Амилоидоз щитовидной железы также своеобразен. Отложения амилоида в строме и сосудах железы могут быть проявлением не только генерализованного амилоидоза, но и медуллярного рака железы (медуллярный рак щитовидной железы с амилоидозом стромы). Амилоидоз стромы часто встречается в опухолях эндокринных органов и APUD-системы (медуллярный рак щитовидной железы, инсулома, карциноид, феохромоцитома, опухоли каротидных телец, хромофобная аденома гипофиза, гипернефроидный рак), причем в образовании APUD-амилоида доказано участие эпителиальных опухолевых клеток. |