Рассеянный склероз 1. Рассеянный склероз
 Скачать 92.5 Kb. Скачать 92.5 Kb.
|

| |||||||||||||||||||||||||||||||||||||||
| Неизвестный антиген (вирус?) | ||
| | 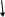 | |
| Активация Т-лимфоцитов | ||
| | | |
| Прохождение через ГЭБ | ||
| | | |
| Реакция Ag - At | ||
| | | |
| Высвобождение цитокинов (интерферон γ и TNF- γ) | ||
| | | |
| Повреждение олигодендроцитов и миелина | ||
| | | |
| Демиелинизация ЦНС | ||
Высокий риск РС ассоциируется со следующими факторами:
внешней среды – ретровирусы, вирус Эпштейна-Бара, герпеса, краснухи;
генетические – HLA-ассоциации: 1 кл. Ag – А3; В7;
2 кл. Ag – DRW15, DW2, DQW6
Клиническая картина.
Начальные проявления РС характеризуются широким спектром неврологических симптомов. Наиболее частые начальные симптомы – это слабость и нарушение чувствительности в одной или более конечностях, снижение зрения на один глаз, нарушение мочеиспускания, мозжечковая атаксия.
Основные клинические проявления:
1. центральные парезы (параличи) конечностей, чаще нижний спастический парапарез;
2. отсутствие брюшных рефлексов;
3. мозжечковая атаксия;
4. побледнение височных половин дисков зрительных нервов, ретробульбарный неврит;
5.нарушение чувствительности по проводниковому типу, парастезии, расстройство вибрационной, реже мышечно-суставной чувствительности, гипалгезия;
6.нарушение функций тазовых органов;
7.неврозоподобные состояния, эйфория.
Классификация рассеянного склероза.
(выделяют следующие формы заболевания)
По уровню поражения:
- церебральная
- спинальная
- цереброспинальная
2. По течению:
- ремитирующая
- первично-прогрессирующая
- вторично-прогрессирующая
Диагностика.
Точная диагностика РС крайне важна, т.к. это заболевание часто скрывается под масками другой патологии ЦНС. Для подтверждения диагноза РС проводятся следующие инструментальные и лабораторные исследования: МРТ, исследование вызванных потенциалов и цереброспинальной жидкости, исследование иммунного статуса (угнетение клеточного иммунитета, повышенное содержание ЦИК в сыворотке крови) и HLA-типирование (наличие антигенов А-3, В-7, DR,DW.)
Диагностические критерии (Poser C.H., 1983)
РС очевиден, если:
А). 2 атаки, 2 клинических уровня поражения ЦНС, один очаг на МРТ;
Б). 2 атаки, один клинический уровень в ЦНС, один очаг на МРТ (клинический и МРТ очаги не совпадают).
Диагностические критерии (МакДональда, 2000)
А). 2 атаки, два объективно обнаруженных очага, дополнительные данные не нужны;
Б). 2 атаки, один объективно обнаруженный очаг, диссеминация в пространстве на МРТ;
В). 1 атака, два объективно обнаруженных очага, диссеминация во времени по МРТ;
Г). 1 атака, один объективно обнаруженный очаг, диссеминация в пространстве на МРТ, либо положительный результат анализа ликвора и 2 и более МРТ очагов типичных для РС;
Дифференциальный диагноз.
Многие заболевания ЦНС на ранних стадиях своего течения протекают с симптоматикой, характерной для разных форм РС: системная красная волчанка, синдром антифосфолипидных антител, синдром Шегрена, первичный васкулит, саркоидоз, болезнь Лайма, тропический спастический парапарез, подострый склерозирующий панэнцефалит, наследственные и идиопатические спиноцеребелярнные синдромы, новообразования ЦНС, паранеопластические синдромы.
Формулировка диагноза по МКБ-10: G35 Рассеянный склероз.
Лечение рассеянного склероза.
I.Лечение в период обострения (не влияет на общее течение заболевания)
Кортикостероиды
- Метилпреднизолон 0,5-1,0 г/день в/в кап № 3-5
с переходом на прием per os 60мг/день – 3 дня, с последующим снижением дозы на 10мг каждые 3 дня
2. Плазмаферез
II.Лечение, влияющее на общий курс заболевания при ремитирующем
течении:
Интерфероны:
- интерферон бета1b (бета-ферон) 250мкг (8млнМЕ) ч/з день, ориентировочная продолжительность лечения 3 года
- интерферон бета1а –ребиф п/к 3р в неделю 22 или 44мкг (6-12млн МЕ)
- авонекс в/м 1 р в неделю 6млн МЕ
2. Копаксон (глатирамера ацетат) – 20мг/день п/к (в/м) 6-24 месяца
3. Иммуноглобулин (сандоглобин) в/в
III. Лечение прогрессирующих форм:
Интерферон бета1b
Цитостатики ( азатиоприн, метотрексат, митоксантрон)
IV.Симптоматическое лечение:
1.При спастичности мышц – баклофен, тизанидин, диазепам
2.При общей слабости – амантадин
3.При дисфункции мочевого пузыря:
-при гиперрефлексии детрузора (низкий остаточный объем)- оксибутин, толтеродин, трициклические антидепрессанты, баклофен.
- при диссенергии детрузора (высокий остаточный объем) – периодическая катетеризация в сочетании с приемом оксибутина или толтеродина, прозерин, электростимуляция мочевого пузыря.
4.При инфнкции мочевыводящих путей – короткий курс антибиотиков
5.При пароксизмальных состояниях – карбамазепин
ТЕМА: Боковой амиотрофический склероз (болезнь двигательного нейрона).
Хроническое дегенеративное заболевание с поражением двигательных нейронов передних рогов спинного мозга, мозгового ствола (периферических двигательных нейронов) и корковых (центральных) двигательных нейронов.
БАС представляет собой спорадическое заболевание, в 10% случаев имеет семейный характер (передается по аутосомно- доминантному типу). В части семейных случаев выявлена мутация на длинном плече 21 хромосомы – в гене, кодирующим фермент супероксиддисмутазу – 1. Для объяснения патогенеза заболевания предложено несколько теорий, ведущими из которых являются: наследственно обусловленная недостаточность Cu – Zn – зависимой супероксиддисмутазы и теория глутаматной эксайтотоксичности. Макроскопические изменения головного и спинного мозга больных, погибщих от БАС, представлены слабовыраженной атрофией. Микроскопически выявлется существенное уменьшение числа центральных и периферических мотонейронов (передние рога спинного мозга, двигательные ядра ствола головного мозга, пирамидные клетки). В большей степени страдают альфа-мотонейроны сегментов шейного утолщения спинного мозга. Выявляются умеренные изменения нейронов ретикулярных ядер ствола мозга.
Классификация БАС.
Спорадический классический (форма Шарко).
Семейный (генетическидетерминированный).
Синдром «БАС – плюс».
БАС начинается в возрасте 50 – 70 лет (только в 5% заболевание развивается до 30 лет) с постепенно нарастающей слабости, неловкости и похудания в дистальных отделах рук. В большинстве случаев симптоматика асимметрична. При осмотре обнаруживаются фибрилляции и фасцикуляции. Со временем слабость и похудание распространяются на более проксимальные мышцы, присоединяются признаки поражения центральных двигательных нейронов – оживление сухожильных рефлексов, патологические стопные и кистевые рефлексы, умеренная спастичность. Параллельно развиваются дизартрия, дисфагия, атрофия и фасцикуляции в языке, парез мягкого неба, которые являются следствием сочетания бульбарного и псевдобульбарного параличей. Постепенно в процесс вовлекаются дыхательные мышцы, в частности диафрагма, что проявляется парадоксальным дыханием (на вдохе живот западает, на выдохе – выпячивается) и развитием дыхательной недостаточности.
Клиническая классификация заболевания двигательного нейрона:
(в зависимости от начального преимущественного поражения центрального или периферического мотонейрона и локализации пораженной мускулатуры)
1. Прогрессирующая мышечная атрофия (поражение по периферическому типу мышц конечностей, чаще верхней, вначале с одной стороны)
2. Прогрессирующий бульбарный паралич (у 20% больных начало с бульбарного паралича)
3. Первичный боковой склероз (наиболее редкий вариант в виде начального спастического парапареза)
4. Боковой амиотрофический склероз (комбинация поражения периферического и центрального мотонейронов на любом уровне, типично для всех больных на поздней стадии заболевания)
Не характерными для БАС являются глазодвигательные и тазовые расстройства. Интеллект обычно сохранен, но у 10% больных, чаще в семейных случаях, развивается деменция, вызванная сопутствующей дегенерацией лобной коры. Средняя длительность жизни после начала заболевания 3 – 4 года, редко - дольше 5 – 7 лет.
Диагноз подтверждается:
Сочетанием признаков поражения периферических и центральных двигательных нейронов.
Сочетанием признаков бульбарного и псевдобульбарного параличей.
Быстрым прогрессированием и развитием летального исхода в течение нескольких лет.
Отсутствием глазодвигательных нарушений, тазовых нарушений и других проявлений вегетативной недостаточности, нарушений зрения и общей чувствительности.
Данными ЭНМГ, которая выявляет денервационные изменения, связанные с поражением клеток передних рогов.
МРТ, КТ (для исключения другой патологии) .
Молекулярно-генетический анализ (мутация СОД-1).
Диагностические критерии.
Согласно пересмотренным Эль-Эскориальским критериям (1998 год) для постановки диагноза БАС необходимо наличие:
признаков поражения периферического мотонейрона по клиническим, электрофизиологическим и патоморфологическим данным;
признаков поражения центрального мотонейрона по клиническим данным, а также прогрессирующего распространения симптомов в пределах одной или нескольких областей иннервации.
Наряду с этим для постановки диагноза БАС необходимо: отсутствие электрофизиологических и патологических признаков другого заболевания, данных нейровизуализации о наличии других заболеваний, которые могли бы объяснить клинические и электрофизиологические признаки.
Формулировка диагноза по МКБ-10: G 12.2-болезнь двигательного нейрона: семейная болезнь двигательного нейрона, боковой амиотрофический склероз, первичный боковой склероз, прогрессирующий бульбарный паралич, прогрессирующая мышечная дистрофия.
Заболевания, с которыми следует дифференцировать БАС: спондилогенная шейная миелопатия, опухоль краниовертебрального перехода, краниовертебральные анамалии, сирингомиелия, спинальные амиотрофии, интоксикация свинцом, ртутью, марганцем, паранеопластический синдром, лимфома, тиреотоксикоз.
Лечение:
Эффективного лечения на сегодняшний день не существует.
Больной и его родственники нуждаются в психологической поддержке.
Рилузол – ингибитор пресинаптического высвобождения глутамата- 100-200мг/д в течение 18 месяцев
Симптоматическое лечение:
- при дисфагии – черезкожная эндоскопическая гастростомия, назо-гастральный зонд
- при избыточной саливации- амитриптилин 25-100мг/д
- при спастичности мышц- миорелаксанты
- ортопедические приспособления
- при респираторных нарушениях – антибактериальная терапия, не допускать аспирации (слюна, пища), вопрос о трахеостомии и респираторной поддержке решается индивидуально