Устойчивость коллоидных систем 2 вида устойчивости флокуляция, коалесценция, структурообразование
 Скачать 0.81 Mb. Скачать 0.81 Mb.
|

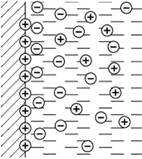
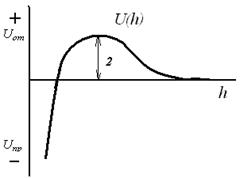
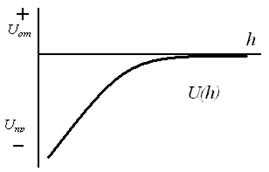
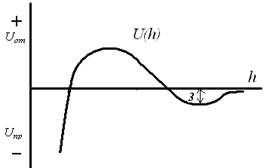
5-10 нм и экспоненциально падает с увеличением толщины. Между лиофобными поверхностями "структурное" притяжение экспериментально обнаруживается на расстояниях не более 80 нм. 1. Для начала коагуляции золя необходима некоторая минимальная концентрация электролита, называемая порогом коагуляции ϒ. 2. Коагулирующим действием обладает тот из ионов электролита, заряд которого противоположен заряду коллоидных частиц, причем коагулирующее действие иона тем сильнее, чем больше его заряд — правило Шульце Гарди или правило значности. Величины порогов коагуляции двухзарядных ионов примерно на порядок, а трехзарядных — на два порядка меньше, чем для однозарядных ионов. Правило значности имеет приближенный характер и справедливо только для неорганических ионов; некоторые однозарядные органические ионы обладают более сильным коагулирующим действием, чем двухзарядные неорганические ионы, что обусловлено их сильной специфической адсорбируемостью. 3. В рядах неорганических ионов с одинаковыми зарядами коагулирующее действие возрастает с уменьшением гидратируемости ионов; например, в ряду однозарядных катионов щелочных металлов коагулирующее действие возрастает от лития к рубидию: 4. В осадках, получаемых при коагуляции золей электролитами, всегда присутствуют ионы, вызвавшие коагуляцию. 5. При коагуляции золей смесями электролитов сравнительно редко наблюдается их независимое (аддитивное) действие; обычно имеет место взаимное усиление либо ослабление коагулирующего действия (синергизм либо антагонизм ионов). Адсорбционная составляющая расклинивающего давления Пa связана с перераспределением концентраций молекул (или ионов) по толщине пленки при перекрывании диффузных адсорбционных слоев. Если перекрываются адсорбционный слои ПАВ или полимеров, возникает стерическая составляющая расклинивающего давления, близкая к адсорбционной. Учет расклинивающего давления и интерпретация его составляющих необходимы при расчете равновесия и устойчивости коллоидных систем (в частности, теория Дерягина-Ландау-Фервея-Овербека учитывает Пт и Пa), пен, свободных и смачивающих тонких пленок, для анализа таких явлений, как полимолекулярная адсорбция, смачивание, флотация. Большинство поверхностных явлений в той или иной степени зависит от различных составляющих расклинивающего давления. 7. Электростатическая составляющая расклинивающего давления; 1. Молекулярная составляющая расклинивающего давления ( 2. Электростатическая составляющая расклинивающего давления ( 3. Адсорбционная составляющая расклинивающего давления ( 4. Структурная составляющая расклинивающего давления ( Таким образом Структурная и адсорбционная составляющие расклинивающего давления В теории устойчивости ДЛФО для лиофобных коллоидных систем адсорбционную и структурную составляющие расклинивающего давления не учитывают и рассматривают только баланс (соотношение) сил притяжения и отталкивания, которые действуют между мицеллами лиофобного золя. Таким образом, для лиофобных коллоидных систем: Зная 8. Молекулярная составляющая расклинивающего давления; 1. Молекулярная составляющая расклинивающего давления ( 2. Электростатическая составляющая расклинивающего давления ( 3. Адсорбционная составляющая расклинивающего давления ( 4. Структурная составляющая расклинивающего давления ( Таким образом Структурная и адсорбционная составляющие расклинивающего давления В теории устойчивости ДЛФО для лиофобных коллоидных систем адсорбционную и структурную составляющие расклинивающего давления не учитывают и рассматривают только баланс (соотношение) сил притяжения и отталкивания, которые действуют между мицеллами лиофобного золя. Таким образом, для лиофобных коллоидных систем: Зная 9. Cуммарная потенциальная энергия взаимодействия частиц в зависимости от расстояния; Взаимодействие двух частиц дисперсной фазы характеризуют с помощью потенциальных кривых – зависимостей энергий взаимодействия между частицами от расстояния (рис. 5.6). В соответствии с теорией ДЛФО соотношения (5.18)–(5.22) определяют поведение дисперсных систем, их устойчивость или скорость коагуляции зависят от знака и значения общей потенциальной энергии взаимодействия частиц U(h). На малых (h → 0) и на больших расстояниях при (h > 200 нм) преобладает энергия взаимного притяжения, а на средних расстояниях – энергия электростатического отталкивания. В результате геометрического сложения этих двух кривых (потенциальных кривых притяжения и отталкивания) получается результирующая кривая (U(h)) – полная энергия системы (суммарная энергия взаимодействия). На потенциальной кривой суммарной энергии взаимодействия можно выделить три участка: 1 – область первичного минимума – непосредственное слипание частиц. Коллоидная система с частицами, находящимися друг от друга на малых расстояниях коагулирует в результате ближнего взаимодействия. Осадки получаются плотными и необратимыми, т.к. энергия притяжения намного превышает энергию отталкивания. 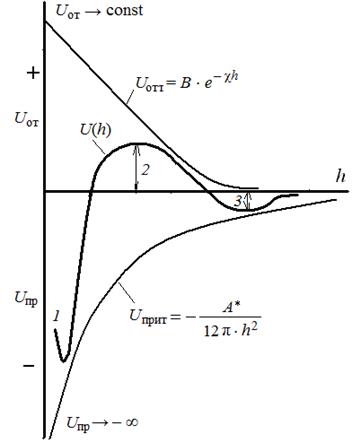 Рис. 5.6. Зависимость энергии притяжения, отталкивания и суммарной энергии взаимодействия частиц от расстояния h 3 – область вторичного минимума – притяжение частиц через прослойку среды. Коллоидная система коагулирует в результате дальнего взаимодействия, осадки получаются рыхлыми и обратимыми, т.к. минимум не глубокий. Вторичному минимуму соответствует явление флокуляции или образование коагуляционных структур. 2 – область с преобладанием сил отталкивания между мицеллами – система агрегативно устойчива – наличие потенциального барьера, препятствующего слипанию частиц. Нарушить эту устойчивость (снизить потенциальный барьер) можно двумя путями: путем повышения температуры, при этом происходит повышение кинетической энергии частиц, что приведет к увеличению числа столкновений; добавить в систему электролит, при этом произойдет сжатие ДЭС, в результате чего частицы могут подойти друг к другу на меньшие расстояния, где усиливаются силы притяжения. Для того чтобы ответить на вопрос об устойчивости дисперсной системы необходимо оценить высоту потенциального барьера (2) и глубину вторичного минимума (3) и сравнить с энергией броуновского движения kT. Рассмотрим следующие случаи:
взаимодействуют друг с другом через прослойку среды и совершают совместное броуновское движение. К паре частиц могут присоединяться на дальних расстояниях другие частицы с образованием более сложных структур. Происходит структурирование золя, при этом дисперсность, удельная поверхность и свободная поверхностная энергия системы не изменяются. Установлено, что в результате дальнего взаимодействия образуются периодические коллоидные структуры. Дальнее взаимодействие между частицами часто наблюдается в золях оксидов и гидроксидов, именно такие системы способны к пептизации. 10. Изменение состояния системы при возрастании концентрации электролита; Если в коллоидный раствор медленно добавлять электролит, то первые его порции не влияют на золь. При увеличении концентрации электролита начинается образование частиц низших порядков (II, III и т.д.), которое протекает незаметно для невооружённого глаза и поэтому называется скрытой коагуляцией. Дальнейшее увеличение концентрации электролита ведёт к прогрессивному развитию процесса коагуляции, повышению её скорости и сопровождается появлением частиц более высоких порядков. Золь претерпевает видимые изменения: он мутнеет или изменяется его окраска. При этом величина ζ-потенциала частиц уменьшается. Эта стадия процесса называется явной коагуляцией. Переход скрытой коагуляции в явную называется порогом коагуляции: ему соответсвует пороговая концентрация электролита, т.е. минимальная концентрация электролита, вызывающая явную коагуляцию. (Измеряется эта величина миллимолях на литр золя.) В это время ζ-потенциал ещё сохраняется, но он обычно не превышает 30 мв и называется критическим ζ-потенциалом. Однако изменение величины ζ-потенциала не всегда соответствует процессу коагуляции частиц. Нередко коагуляция начинается при высоких значениях ζ-потенциала, а иногда с понижением этого потенциала некоторые золи даже увеличивают свою устойчивость. Это подтверждпет, что ζ-потенциал является важным, но не единственным определяющим фактором устойчивости коллоидных частиц. 11. Лиофильные дисперсные системы; Лиофильные- это системы, частицы дисперсной фазы которых интенсивно взаимодействуют с дисперсионной средой К лиофобным системам относятся золи драгоценных металлов, золи металлоидов (серы, селена, теллура), дисперсии полимеров в воде (например, полистирола, фторолона), золи сульфидов мышьяка, сурьмы, кадмия, ртути, золи гидроксидов железа, алюминия и т.д. Эти системы характеризуются, так называемой, кинетической устойчивостью и агрегативной неустойчивостью и требуют стабилизации. К лиофильным коллоидным системам Фрейндлих отнес растворы, образующиеся при растворении природных или синтетических ВМС. Таковы растворы белков, крахмала, пектинов, камедей, эфиров целлюлозы и разнообразных смол, как природных так и синтетических. 12. Классификация ПАВ; По способности к диссоциации в водных растворах ПАВ делятся на ионогенные (анионные, катионные и амфотерные) и неионогенные. Анионные ПАВ диссоциируют в воде с образованием поверхностно-активного аниона. К анионным ПАВ относятся карбоновые кислоты и соли синтетических жирных кислот (стеарат натрия, олеат натрия), алкилсульфаты, алкиларилсульфонаты, лауретсульфаты, сульфосукцинаты и другие типы поверхностно-активных анионов (фосфаты, тиосульфаты). Алкилсульфаты и алкиларилсульфонаты являются сильными кислотами и могут быть использованы в кислых и солевых растворах (в отличие от солей жирных кислот имеющих низкую эффективность в кислых средах). Катионные ПАВ диссоциируют в воде с образованием поверхностно-активного катиона. С помощью катионных ПАВ стабилизируют дисперсные системы с получением положительно заряженных частиц. Катионные ПАВ используются в качестве бактерицидных и дезинфицирующих веществ, ингибиторов коррозии (Cublen применяется в промышленной и автохимии). Амфотерные ПАВ имеют две функциональные группы. В зависимости от рН среды обладают анионактивными или катионактивными свойствами. В щелочной среде проявляют анионактивные свойства, в кислой среде — катионактивные. К амфотерным ПАВ относятся бетаины, аминоксиды и такие ПАВ как имидазолины (кокоамфодиацетат натрия). Неионогенные ПАВ не диссоциируют в растворах на ионы, являются смесью гомологов с различной длиной полиоксиэтиленовой цепи (этоксилированные жирные кислоты, амиды жирных кислот, оксиэтилированные алкилспирты) Химические свойства неионогенных ПАВ легко регулировать, изменяя длину полиоксиэтиленовой цепи. Они могут использоваться как в присутствии растворимых солей, так и в кислой или щелочной средах. Пенообразующая способность ионогенных ПАВ существенно выше, чем неионогенных ПАВ. Это связано с большей скоростью образования адсорбционных слоев у ионогенных ПАВ. Стабильность пены повышается с ростом концентрации. 13. Критическая концентрация мицеллообразования; ККМ - это концентрация ПАВ, при достижении которой при добавлении ПАВ в раствор концентрация на границе раздела фаз остается постоянной, но в то же время происходит самоорганизация молекул ПАВ в объёмном растворе (мицеллообразование или агрегация). В результате такой агрегации образуются так называемые. Отличительным признаком мицеллообразования служит помутнение раствора ПАВ. Водные растворы ПАВ, при мицеллообразовании также приобретают голубоватый оттенок (студенистый оттенок) за счёт преломления света мицеллами. Все методы определения ККМ основаны на регистрации излома на графике, показывающем концентрационную зависимость физикохимических свойств растворов ПАВ (поверхностного натяжения сг, мутности г, молярной электрической проводимости Я, осмотического давления п, показателя преломления п (рис. 5.3). Одна из ветвей таких кривых описывает свойства системы в молекулярном состоянии, другая — в коллоидном. Точку перелома считают соответствующей переходу молекул в мицеллы, то есть соответствующей ККМ.  Рис. 5.3. Зависимость физико-химических величин от концентрации ПАВ 14. Гидрофильно-липофильный баланс; прямые и обратные мицеллы; Свойства ПАВ определяются количественным соотношением гидрофильных и липофильных групп в его составе (ГЛС) или гидрофильно-липофильным балансом (ГЛБ). Впервые количественное значение ГЛБ было предложено и эмпирически определено Гриффином [2,3] для описания эффективности действия ПАВ в качестве эмульгаторов первого или второго рода. ( Эмульгатор первого рода стабилизирует эмульсии масло/вода, эмульгатор второго рода является стабилизатором эмульсии вода/масло). Система ГЛБ имеет шкалу от 0 до 40; ПАВ с выраженными липофильными свойствами (растворимые в органических растворителях) имеют низкие значения ГЛБ, а гидрофильные вещества — высокое. Шкала Гриффина с самого начала была рассчитана на практическое применение (подбор нужного эмульгатора) и получила широкое распространение [20Р]: наличие у каждого ПАВ своего числа ГЛБ позволяло составлять без труда рецепты смесей ПАВ с нужным числом ГЛБ. Ниже приводятся примерные пределы чисел ГЛБ и соответствующие им применения ПАВ [0-12А]: 3,5—6 Эмульгаторы второго рода 7—9 Смачиватели 8— 18 Эмульгаторы первого рода 13—15 Моющие агенты (детергенты) 15—18 Солюбилизаторы Чтобы найти число ГЛБ для конкретного ПАВ, необходимо испытать его в сравнении с уже известными веществами и найти ему место в пределах шкалы. Попытка создания теоретической базы ГЛБ была предпринята Дэвисом. Он подобрал так называемые групповые числа, т. е. числа ГЛБ не для молекул в целом, а для составляющих их групп (структурных единиц молекулы). При сложении этих чисел получается ГЛБ ПАВ по формуле: ГЛБ = Σ гидрофильных групповых чисел + + Σ гидрофобных групповых чисел + 7 15. Влияние на ККМ природы ПАВ; Влияние температуры на ККМ ионогенных ПАВ и неионогенных ПАВ различно. Повышение температуры приводит к увеличению ККМ ионогенного ПАВ из-за дезагрегирующего действия теплового движения, а также к уменьшению мицелл [47]. Повышение температуры приводит к уменьшению ККМ неионогенного ПАВ за счет дегидратации оксиэтиленовых цепочек (неионогенные ПАВ всегда образованы полиоксиэтиленовыми цепочками и углеводородными «хвостами»). На величину ККМ влияют: • строение и длина углеводородной цепи; • характер полярной группы; • наличие в растворе индифферентных электролитов и неэлектролитов; • температура. 16. Влияние на ККМ природы электролита и температуры; Влияние температуры на ККМ определяется взаимодействием различных факторов, и характер его различен в случаях ионогенных и неионогенных ПАВ. Вообще повышение температуры должно затруднять образование мицелл вследствие возрастания дезагрегирующего влияния теплового движения молекул. Вместе с тем с увеличением интенсивности теплового движения уменьшается гидратация полярных групп молекул (ионов) ПАВ, что, напротив, способствует мицеллообразованию. Важную роль играют структурные изменения воды при нагревании. Повышение температуры вызывает разупорядочение воды, а это означает, что усиливается стремление воды «избавиться» от гидрофобных частиц (радикалов молекул ПАВ), которые, как уже отмечалось, способствуют ее структурированию. Таким образом, происходит усиление гидрофобных взаимодействий в воде при повышении температуры, что проявляется в снижении величины ККМ. На рисунке 18 приведена типичная картина влияния температуры на ККМ (и мицеллярную массу) неионогенных и ионогенных ПАВ. Величина ККМ НПАВ заметно понижается с повышением температуры, тогда как ККМ ионогенных ПАВ слабо зависит от температуры, незначительно повышаясь при нагревании. Гидрофильные свойства НПАВ обусловлены наличием в полиоксиэтиленовой цепи гетероатомов кислорода (и концевых полярных групп — ОН). Дегидратация полиоксиэтиленовых цепочек при повышении температуры  Рис. 18 Зависимость ККМ (а) и мицеллярной массы (б) от температуры для ионогенных и неионогенных ПАВ: 17. Строение и форма мицелл; Рис 3.1. Схема строения сферической мицеллы ионного ПАВ в воде .  18. Рост мицелл; 19. Термодинамика образования мицелл; Две модели мицеллообразования. 1)Двухфазная(псевдофазная) модель. Мицеллообразование рассматривается как граница раздела фаз. 2)Химическое равновесие- Мицеллообразование рассматривается как обратимая химическая реакция m(ПАВ)<=>(ПАВ)m. время жизни (ПАВ)m=10-7сек. Образование прямых мицелл  Сферическая мицелла Гартли, число агрегации 20-100 Сферическая мицелла Гартли, число агрегации 20-1000>ΔG=ΔH-TΔS-В водной среде, процесс самопроизвольный. ΔH- не велика и может быть положительной.  Н2О- структурированная жидкость, при введении в Н2О неполярных частиц происходит упорядочивание воды около этих неполярных частиц. Это явление термодинамически не выгодно, поэтому образованные водородные связи рвутся и возрастает ΔSН2О. Движущей силой мицеллообразования является положительная энтропия. ΔSсист=ΔSмиц↓+ ΔSН2О↑-энтропия системы. Образование обратных мицелл( числа агрегации 30-40) Н2О- структурированная жидкость, при введении в Н2О неполярных частиц происходит упорядочивание воды около этих неполярных частиц. Это явление термодинамически не выгодно, поэтому образованные водородные связи рвутся и возрастает ΔSН2О. Движущей силой мицеллообразования является положительная энтропия. ΔSсист=ΔSмиц↓+ ΔSН2О↑-энтропия системы. Образование обратных мицелл( числа агрегации 30-40)образуются в неводных средах. Движущая сила- взаимодействие полярных групп между собой. TRlnKKM=A-Bn Число агрегации- число молекул ПАВ входящих в систему. 20. Солюбилизация; влияние различных факторов на солюбилизацию;  21. Механизм солюбилизации. Солюбилизация сопровождается равновесным распределением вещества солюбилизата между водной фазой и мицеллярной. Поэтому процесс коллоидного растворения в мицеллах ПАВ условно можно разделить на следующие стадии: ― растворение солюбилизата в воде; ― диффузия его молекул из объѐма раствора в мицеллы ПАВ; ― проникновение и распределение солюбилизата внутри мицелл. Процесс солюбилизации является медленным, равновесие может устанавливаться в течение нескольких суток. Перемешивание и повышение температуры интенсифицируют этот процесс. При интенсивном перемешивании лимитирующей будет третья стадия, а скорость солюбилизации будет определяться количеством вакантных мест в мицеллах и факторами, влияющими на структуру сольватных оболочек. Повышение температуры увеличивает истинную растворимость углеводородов в воде, ускоряет диффузию и облегчает проникновение солюбилизата в мицеллы вследствие снижения плотности упаковки мицеллы из-за теплового движения |