лекции. Вопросы лекции
 Скачать 5.13 Mb. Скачать 5.13 Mb.
|

Изотермы адсорбции и форма фронтов зонРассмотрим вопросы применения теории адсорбции к описанию хроматографических разделений. Основными задачами теории адсорбции в приложении к хроматографии являются получение ответов на два вопроса: в какой последовательности зоны разделяемых веществ будут выходить из хроматографической колонки; какие условия в ходе процесса разделения необходимо соблюдать, чтобы фронты зон разделяемых компонентов были обостренными. Процессы хроматографических разделений могут рассматриваться либо с позиций линейной и идеальной, либо с позиций нелинейной и неидеальной теории. Линейная и идеальная теория является упрощенным вариантом нелинейной и неидеальной теории, поскольку предполагает существование следующих допущений: адсорбционное равновесие в процессе разделения устанавливается мгновенно, т.е. сорбционные связи устанавливаются мгновенно; процессом продольной диффузии разделяемых соединений в колонке можно пренебречь, т.е. перемешивание зон разделяемых компонентов не обусловлено процессом продольной диффузии. 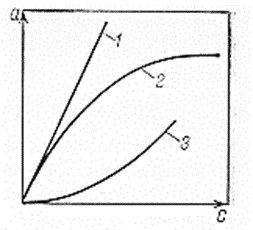 Рисунок 5. Рисунок 5.Эти положения конечно же не соответствуют реальному эксперименту. Для установления адсорбционных связей всегда требуется некоторое время, необходимое для диффузии молекул разделяемых соединений из подвижной фазы к поверхности адсорбента, время на диффузию этих молекул от поверхности зерна к центру, и наконец, время на установление адсорбционных связей. Далее, в потоке подвижной фазы всегда имеет место процесс перемешивания зон разделяемых компонентов, т.е. существованием процесса продольной диффузии пренебрегать также нельзя. Нелинейная и неидеальная теория обязательно учитывает эти факторы процесса разделения. Однако для предварительных рассуждений вполне можно ограничиться представлениями линейной и идеальной теории, поскольку с технической стороны возможно организовать выполнение экспериментальных исследований таким образом, чтобы достаточно строго реализовать требования нелинейной и неидеальной теории: использовать для разделений адсорбент с очень малым диаметром гранул; использовать для разделений очень маленькую скорость подвижной фазы. Эти условия эксперимента будут способствовать очень быстрому установлению адсорбционного равновесия и существенному снижению влияния процессов продольной диффузии на перемешивание зон разделяемых компонентов. Таким образом, выводы линейной и идеальной теории оказываются очень вескими, поскольку если они предсказывают невозможность разделения компонентов данной смеси, то кинетические факторы нелинейной и неидеальной теории будут только усиливать эту невозможность. Если линейная и идеальная теория предсказывает возможность разделения, целесообразно попробовать реализовать эту возможность практически. В этой связи рассмотрим, что дают представления линейной и идеальной теории с точки зрения обеспечения оптимальных условий для обострения фронтов зон разделяемых веществ. Используем полученное ранее выражение для описания скорости перемещения фронта зоны по колонке: 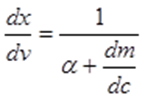 ( 23) ( 23)Приведенное уравнение позволяет сделать весьма важный вывод – скорость перемещения фронта зоны, а следовательно, и оптимизация условий обострения фронтов зоны определяется видом изотермы адсорбции. Рассмотрим вопросы формирования фронтов зон в случае описания процесса адсорбции изотермой Лэнгмюра. Верхняя часть изотермы адсорбции, описывающая область высоких равновесных концентраций, соответствует головному, или переднему, фронту зоны. Нижняя часть изотермы, область низких равновесных концентраций, соответствует заднему фронту - хвостовой части зоны. При хроматографическом разделении непрерывно, последовательно повторяется один и тот же элементарный акт адсорбции и десорбции и исследуемое соединение либо полностью находится в адсорбенте (адсорбция), либо полностью находится в объеме подвижной фазы (десорбция). Величина для плотно упакованных колонок имеет очень малое численное значение и остается постоянной в течение всего процесса разделения. Следовательно, для головной части изотермы адсорбции производная Таким образом, молекулы, входящие в головную часть зоны, продвигаются быстро и с одинаковой скоростью, что благоприятствует созданию условий обострения фронта зоны. Для хвостовой части зоны величина производной Таким образом, если процесс адсорбции описывается уравнением мономолекулярной адсорбции Лэнгмюра, то на хроматограмме передний фронт зоны обострен, а задний фронт – размыт. К этому выводу можно прийти и при установлении физического смысла связи изотермы адсорбции и размывания фронтов зоны. На начальном участке изотермы адсорбции, в области малых равновесных концентраций, хвосте зоны, на поверхности адсорбента много свободных адсорбционных центров, коэффициент распределения молекул исследуемого соединения, определяемый отношением концентрации в адсорбенте к концентрации в подвижной фазе - велик. В результате этого скорость перемещения молекул мала и различна для различных участков зоны. Головная часть зоны соответствует участку изотермы полного насыщения адсорбционных центров, силы адсорбции уменьшены и скорость перемещения молекул высокая и одинаковая. В случае изотермы полимолекулярной адсорбции Фрейндлиха для головной части зоны величины производных Рассмотрим третий случай, когда изотерма адсорбции линейна во всех областях равновесных концентраций. В этом случае величина производной Из приведенного материала следует весьма важный вывод, позволяющий реализовать максимальную эффективность разделения используемой хроматографической колонки. Поскольку следует стремиться к таким условиям процесса разделения, когда пики на хроматограмме регистрируются как симметричные, реализовать это возможно лишь в тех случаях, когда величины равновесных концентраций разделяемых соединений в подвижной фазе соответствуют закону Генри, т.е. располагаются на начальных линейных участках изотерм адсорбции. 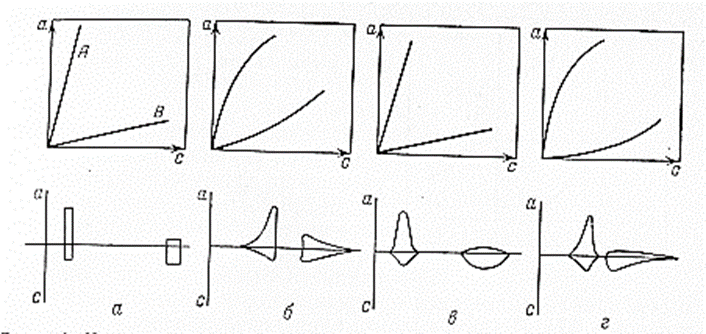 Рисунок 6. Соответствие пик на хроматограмме типам изотерм адсорбции   Изотермы адсорбции и форма фронтов зон Рассмотрим вопросы применения теории адсорбции к описанию хроматографических разделений. Основными задачами теории адсорбции в приложении к хроматографии являются получение ответов на два вопроса: в какой последовательности зоны разделяемых веществ будут выходить из хроматографической колонки; какие условия в ходе процесса разделения необходимо соблюдать, чтобы фронты зон разделяемых компонентов были обостренными. Процессы хроматографических разделений могут рассматриваться либо с позиций линейной и идеальной, либо с позиций нелинейной и неидеальной теории. Линейная и идеальная теория является упрощенным вариантом нелинейной и неидеальной теории, поскольку предполагает существование следующих допущений: адсорбционное равновесие в процессе разделения устанавливается мгновенно, т.е. сорбционные связи устанавливаются мгновенно; процессом продольной диффузии разделяемых соединений в колонке можно пренебречь, т.е. перемешивание зон разделяемых компонентов не обусловлено процессом продольной диффузии. Рисунок 5.Эти положения конечно же не соответствуют реальному эксперименту. Для установления адсорбционных связей всегда требуется некоторое время, необходимое для диффузии молекул разделяемых соединений из подвижной фазы к поверхности адсорбента, время на диффузию этих молекул от поверхности зерна к центру, и наконец, время на установление адсорбционных связей. Далее, в потоке подвижной фазы всегда имеет место процесс перемешивания зон разделяемых компонентов, т.е. существованием процесса продольной диффузии пренебрегать также нельзя. Нелинейная и неидеальная теория обязательно учитывает эти факторы процесса разделения. Однако для предварительных рассуждений вполне можно ограничиться представлениями линейной и идеальной теории, поскольку с технической стороны возможно организовать выполнение экспериментальных исследований таким образом, чтобы достаточно строго реализовать требования нелинейной и неидеальной теории: использовать для разделений адсорбент с очень малым диаметром гранул; использовать для разделений очень маленькую скорость подвижной фазы. Эти условия эксперимента будут способствовать очень быстрому установлению адсорбционного равновесия и существенному снижению влияния процессов продольной диффузии на перемешивание зон разделяемых компонентов. Таким образом, выводы линейной и идеальной теории оказываются очень вескими, поскольку если они предсказывают невозможность разделения компонентов данной смеси, то кинетические факторы нелинейной и неидеальной теории будут только усиливать эту невозможность. Если линейная и идеальная теория предсказывает возможность разделения, целесообразно попробовать реализовать эту возможность практически. В этой связи рассмотрим, что дают представления линейной и идеальной теории с точки зрения обеспечения оптимальных условий для обострения фронтов зон разделяемых веществ. Используем полученное ранее выражение для описания скорости перемещения фронта зоны по колонке: ( 23)Приведенное уравнение позволяет сделать весьма важный вывод – скорость перемещения фронта зоны, а следовательно, и оптимизация условий обострения фронтов зоны определяется видом изотермы адсорбции. Рассмотрим вопросы формирования фронтов зон в случае описания процесса адсорбции изотермой Лэнгмюра. Верхняя часть изотермы адсорбции, описывающая область высоких равновесных концентраций, соответствует головному, или переднему, фронту зоны. Нижняя часть изотермы, область низких равновесных концентраций, соответствует заднему фронту - хвостовой части зоны. При хроматографическом разделении непрерывно, последовательно повторяется один и тот же элементарный акт адсорбции и десорбции и исследуемое соединение либо полностью находится в адсорбенте (адсорбция), либо полностью находится в объеме подвижной фазы (десорбция). Величина для плотно упакованных колонок имеет очень малое численное значение и остается постоянной в течение всего процесса разделения. Следовательно, для головной части изотермы адсорбции производная Таким образом, молекулы, входящие в головную часть зоны, продвигаются быстро и с одинаковой скоростью, что благоприятствует созданию условий обострения фронта зоны. Для хвостовой части зоны величина производной Таким образом, если процесс адсорбции описывается уравнением мономолекулярной адсорбции Лэнгмюра, то на хроматограмме передний фронт зоны обострен, а задний фронт – размыт. К этому выводу можно прийти и при установлении физического смысла связи изотермы адсорбции и размывания фронтов зоны. На начальном участке изотермы адсорбции, в области малых равновесных концентраций, хвосте зоны, на поверхности адсорбента много свободных адсорбционных центров, коэффициент распределения молекул исследуемого соединения, определяемый отношением концентрации в адсорбенте к концентрации в подвижной фазе - велик. В результате этого скорость перемещения молекул мала и различна для различных участков зоны. Головная часть зоны соответствует участку изотермы полного насыщения адсорбционных центров, силы адсорбции уменьшены и скорость перемещения молекул высокая и одинаковая. В случае изотермы полимолекулярной адсорбции Фрейндлиха для головной части зоны величины производных Рассмотрим третий случай, когда изотерма адсорбции линейна во всех областях равновесных концентраций. В этом случае величина производной Из приведенного материала следует весьма важный вывод, позволяющий реализовать максимальную эффективность разделения используемой хроматографической колонки. Поскольку следует стремиться к таким условиям процесса разделения, когда пики на хроматограмме регистрируются как симметричные, реализовать это возможно лишь в тех случаях, когда величины равновесных концентраций разделяемых соединений в подвижной фазе соответствуют закону Генри, т.е. располагаются на начальных линейных участках изотерм адсорбции. Рисунок 6. Соответствие пик на хроматограмме типам изотерм адсорбции 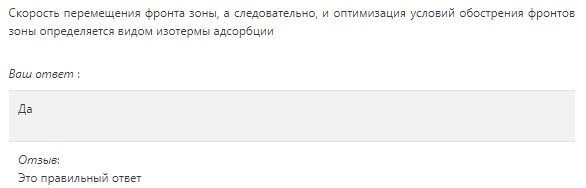 Основные факторы размывания хроматографических пиков Для того чтобы разделить бинарную смесь компонентов, необходимо, чтобы они находились в колонке разное время. Однако даже время пребывания отдельных молекул одного и того же вещества в большей или меньшей степени отличается от среднего значения, характерного для этого вещества. Причиной этому являются процессы диффузии, конвекции и замедленного обмена между подвижной и неподвижной фазами. Насадочные колонки независимо от их внутреннего диаметра представляют собой трубки, заполненные частицами сорбента, которые образуют стационарный зернистый слой. Поток газа фильтруется через этот слой, двигаясь по транспортным каналам, образуемым зазорами между частицами. За счет разных по длине путей перемещения молекул разделяемых соединений возникает специфический размывающий фактор, характеризуемый “вихревой” диффузией.  Рисунок 7. Схема перемещения исследуемых веществ по транспортному каналу Рисунок 7. Схема перемещения исследуемых веществ по транспортному каналу В капиллярных колонках имеется единственный транспортный канал вдоль ее оси. В этой связи в капиллярных колонках “вихревая” диффузия отсутствует, но возникает другой размывающий фактор, связанный с параболическим распределением скоростей по сечению канала, характеризуемый так называемой “тейлоровской” диффузией. Вследствие такого «рассеяния» времени пребывания в колонке отдельных молекул концентрация вещества на выходе из колонки изменяется во времени, при этом профиль концентрации подчиняется уравнению функции нормального распределения ошибок Гаусса, которое характеризует распределение концентрации исследуемого соединения C в пространстве в фиксированный момент времени “х” от времени положения максимума хроматографического пика где Смакс– величина концентрации вещества в точке максимума пика, численное значение которой рассчитывается из уравнения (24) при х = 0 и равная коэффициенту перед экспоненциальным членом уравнения Гаусса Параметр в уравнениях (24) и (25) называется средним квадратичным отклонением, а величину называют дисперсией. Этот параметр характеризует степень размывания кривой распределения случайных ошибок, а в случае хроматографических разделений – ширину регистрируемого хроматографического пика у основания (рис. 1). Чтобы придать величине среднего квадратичного отклонения графическую интерпретацию, допустим, что в уравнении (24) отношение 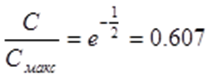 (26) (26)Тогда с учетом уравнения (24) можно записать: Отсюда, приравнивая показатели экспонент, получим х =σ . Это означает, что полуширина хроматографического пика, измеренная на высоте, составляющей 0,607 от максимальной высоты пика, равна среднеквадратичному отклонению . Кривая Гаусса имеет колоколообразную форму: наряду с максимумом она имеет две точки перегиба. Если к этим точкам перегиба провести касательные, то величина отрезка, отсекаемого касательными на оси абсцисс, характеризует ширину хроматографического пика у основания ω и оказывается равной 4 (рис. 8). 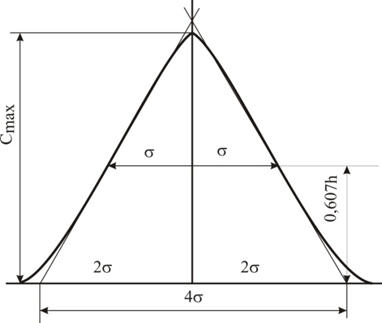 Рисунок 8. Хроматографический пик Рисунок 8. Хроматографический пикК числу регулируемых факторов, влияющих на характер хроматограммы можно отнести: 1. Размер и однородность частиц сорбента; 2. Длину колонки; 3. Скорость газа-носителя; 4. Температуру колонки; 5. Объем вводимой пробы. 1. Увеличение диаметра зерен приводит к увеличению размывания пика. Поэтому следует работать с сорбентом, имеющим минимальную крупность и максимальную однородность по крупности. Чаще всего используют фракции носителя с диаметром зерен 0,15-0,20 и 0,20-0,30 мм. 2. С увеличением длины колонок улучшается разделение. Так, увеличивая длину колонки с 1 м до 4м. можно увеличить критерий разделения в два раза, увеличение же длины от 3 до 4м дает весьма малый выигрыш в разделении. Предел удлинения колонки достигается очень быстро. При использовании носителя с зернением 0,15-0,20 мм колонки длиной выше 5 м при оптимальной скорости газа-носителя имеют на выходе давление 2,5-3,5·105 Па, что препятствует вводу пробы шприцем. 3. Значительное уменьшение скорости газа-носителя может привести к снижению критерия разделения, а увеличение скорости лишь немного уменьшает его значение. 4. Температуру анализа следует подбирать экспериментально; увеличение ее приводит к уменьшению допустимого объема пробы и, соответственно, ширины пика на хроматограмме. 5. Размер введенной пробы должен отвечать двум требованиям, противоречащим друг другу: - проба должна быть настолько малой, чтобы не ухудшалось разделение компонентов; - проба должна быть настолько большой, чтобы при заданных условиях хроматографирования и детектирования количество определяемого вещества можно было измерить. В количественном хроматографическом анализе следует стремиться к получению хроматограмм с гауссовыми пиками. Измерение асимметричных пиков, как правило, проводится с меньшей точностью. 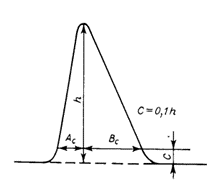 Рисунок. К определению фактора асимметричности хроматографического пика Рисунок. К определению фактора асимметричности хроматографического пикаДля определения принадлежности формы хроматографического пика к гауссовой удобно использовать отнесение ширины пика при основании к полуширине пика. Для истинно гауссовых пиков должно соблюдаться равенство: α = 1,698·а0,5 В условиях реальной газовой хроматоргафии в первом приближении можно считать пик симметричным (гауссовым), если величина а/а0,5 находится в пределах 1,67-1,73. 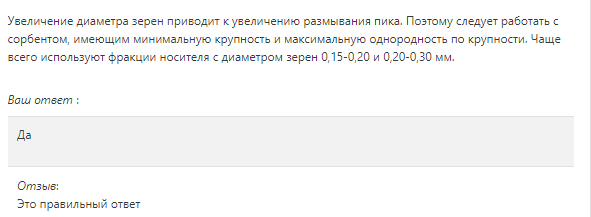 Теория теоретических тарелок Теория теоретических тарелок разработана для описания процесса дистилляции (1952 г , Мартин и Синдж, Нобелевская премия за открытие распределительной хроматографии), однако она является общей для всех многостадийных процессов и позволяет оценить эффективность колонки. Теория теоретических тарелок является формальной и основана на представлении, что хроматографируемое вещество проходит через слой сорбента не непрерывным потоком, а порциями, распределяясь между подвижной и неподвижной фазами на отдельных элементарных участках слоя - так называемых «тарелках». Через каждую такую тарелку вещество проходит периодическими толчками. При этом предполагается, что за время каждого толчка, т. е. практически мгновенно, на тарелках успевает установиться равновесие распределения всех компонентов между подвижной и неподвижной фазами. Таким образом, согласно этой теории, хроматографический процесс является многоступенчатым и состоит из большого числа актов сорбции=десорбции или растворения=испарения компонентов анализируемого вещества в хроматографической колонке, а сама колонка рассматривается как совокупность многих дискретных ступеней - тарелок, хотя в действительности слой адсорбента или пленка неподвижной жидкой фазы в колонке является непрерывным. Анализируемое вещество вместе с элюентом попадает на первую тарелку. Новая порция элюента, подаваемая на первую тарелку, приводит к новому распределению вещества между подвижной и неподвижной фазами, причем часть вещества с данной тарелки переносится на следующую. На этой тарелке также мгновенно устанавливается равновесие, а часть вещества уносится на следующие тарелки. Вследствие этого с каждой новой порцией элюента концентрация вещества на первой тарелке падает, а на последующих возрастает. В результате такого перемещения и перераспределения хроматографируемое вещество оказывается не на одной, а на нескольких тарелках, причем на средних его концентрация достигает максимального значения по сравнению с соседними, так как свежие порции элюента, поступающие в колонку, встречают на первых тарелках все меньшие количества данного компонента в неподвижной фазе. Таким образом, вещество размывается по некоторой толщине слоя неподвижной фазы в колонке, по нескольким тарелкам, причем, чем сильнее размывание, тем большее число тарелок занимает вещество. Следовательно, число тарелок, занимаемых данным компонентом анализируемого вещества, может служить мерой степени размывания вещества по слою адсорбента, мерой эффективности колонки. Такой прием замены реального процесса, протекающего в реальной хроматографической колонке непрерывно и неравновесно, эквивалентным по результатам многоступенчатым дискретным процессом, также приводящим к размыванию полосы компонента, позволил на основании теории скоростей вывести уравнение хроматографической кривой, т.е. дал математическую модель продвижения полосы компонента через колонку. Гауссов характер хроматографического пика обусловлен беспорядочным статистическим характером перемещения большого числа частиц вещества в хроматографической колонке. Одни частицы передвигаются в ней быстрее, другие медленнее, и значения скорости перемещения имеют симметричный разброс относительно среднего значения, характеризующего поведение в колонке некоторой усредненной молекулы.
Если длину слоя сорбента в колонке (длину колонки) L, на которой осуществляется разделение смеси веществ и расположено некоторое число n теоретических тарелок, необходимое для разделения анализируемой смеси веществ, разделить на это число n, то получается величина Н, называемая высотой, эквивалентной одной теоретической тарелке (ВЭТТ): Высота эквивалентной теоретической тарелки представляет собой толщину слоя сорбента, необходимую для установления равновесного распределения вещества между подвижной и неподвижной фазами. Таким образом, число теоретических тарелок n и высота эквивалентной теоретической тарелки Н являются величинами, характеризующими эффективность хроматографической колонки. Высота эквивалентной теоретической тарелки выражают в единицах длины, как правило в миллиметрах. Так как w = 4s мм, экспериментально Н можно определить как дисперсию, приходящуюся на единицу длины колонки L,мм, непосредственно из хроматограммы, используя полученное на хроматограмме значение ширины пика w у его основания для нахождения величины s: 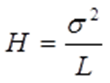 (29) (29)Так как 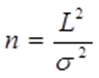 . Приняв время удерживания tR эквивалентом длины колонки, можно установить, что число теоретических тарелок n равно: . Приняв время удерживания tR эквивалентом длины колонки, можно установить, что число теоретических тарелок n равно: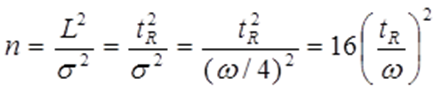 (30) (30)Если ширина пика измерена на середине его высоты, то w1/2 = 2,35 s и 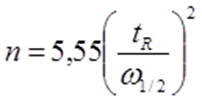 (31) (31)Под эффективностью в хроматографии понимают способность системы "предотвращать" (ограничивать) размывание зон разделяемых веществ. Эффективность колонки тем выше, чем уже пик получается при том же времени удерживания, и измеряется числом теоретических тарелок. Хроматографическая колонка считается высокоэффективной, когда размывание полос небольшое, пики узкие, высота Н составляет 0,3-1 мм. В идеальном случае величина Н приближается к диаметру dp зерна сорбента. При уменьшении значения Н максимумы на хроматограмме становятся более острыми. Для сравнения эффективности двух хроматографических колонок следует использовать приведенную высоту h тарелки: Теория теоретических тарелок позволяет сравнить эффективность различных колонок, оценить качество сорбента и заполнения колонки. Но эта теория не позволяет выявить зависимость эффективности работы хроматографической колонки от скорости подачи подвижной фазы, природы и дисперсности сорбента, не может дать практических рекомендаций, позволяющих минимизировать размывание хроматографических пиков.   |