1. Наследственность и изменчивость
 Скачать 0.68 Mb. Скачать 0.68 Mb.
|

|
Ответы к зачету по мед.генетике. 1. Наследственность и изменчивость. Генетика изучает два неразрывных свойства живых организмов: наследственность и изменчивость, а также методы управления ими. Поэтому именно наследственность и изменчивость являются предметом генетики. Наследственность – это свойство живых организмов передавать свои признаки и особенности развития в неизменном виде следующему поколению. Каждый вид растения и животного сохраняет в процессе размножения характерные для него черты: курица выводит цыплят, свинья рожает поросят, пшеница дает пшеницу и т. д. Если гены принадлежат хромосомам и передаются вместе с ними потомкам, говорят о ядерной наследственности, если же гены входят в состав некоторых клеточных органелл, например, хлоропластов, митохондрий или плазмид, говорят о неядерной наследственности (цитоплазматической). Термин «наследственность» отличается по смыслу от термина наследование. «Наследование» обозначает процесс передачи какого-то конкретного признака от родителей потомству. Например, наследование цвета глаз, формы ушей, цвета кожи и т. д. Наряду с явлением наследственности в предмет исследования генетики входит изучение изменчивости. Изменчивость – это разнообразие в проявлении признаков. Изменчивость заключается в изменении наследственных задатков в процессе их передачи потомству и последующего развития организма. Самым ярким примером изменчивости является разнообразие признаков у человека. Варьирует в потомстве все – морфологические признаки, физиологические, обмен веществ, психика, иммунитет и т. д. Вместе с тем каждый из нас хорошо знает, какие признаки он взял от матери и отца, чем похож на бабушку и дедушку, братьев и сестер. Существует несколько типов изменчивости: наследственная, ненаследственная и онтогенетическая. 2. Предмет и задачи цитогенетики. Цитогенетика - область генетики, изучающая цитологические основы наследственности и изменчивости. Основной предмет исследований цитогенетики включает хромосомы, их организация, функционирование и наследование. Основными задачами цитогенетики является изучение: 1) информационной функции хромосом, т.к. содержащаяся в них ДНК сосредотачивает подавляющую часть различных генов, составляющих геном клетки. 2) транскрипционной функции хромосом, т.к. с них осуществляется считывание генетической информации. 3) структурно-организационной функции, связанной с различными проявлениями генов при изменении структуры хромосом 4) сегрегационной функции хромосом, обеспечивающей распределение аллельных вариантов в различные споры или гаметы при мейозе 5) рекомбинационной функции хромосом, связанной с процессом кроссинговера. 3. Процессы транскрипции и трансляции. 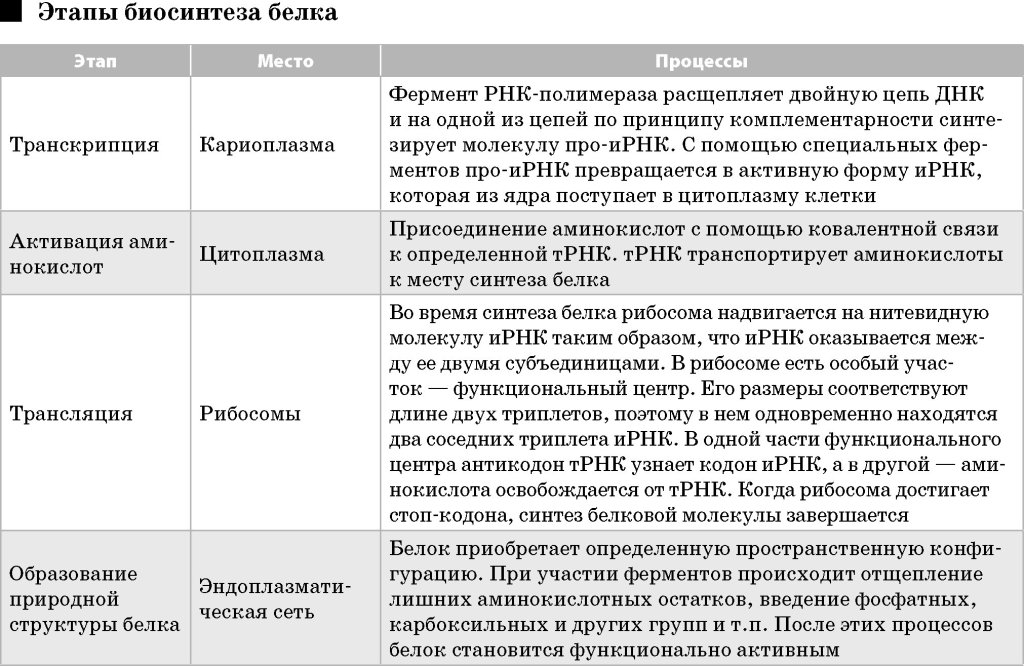 4. Законы Г. Менделя. В первых опытах Г. Мендель принимал во внимание только одну пару признаков. Такое скрещивание носит название моногибридного. После анализа результатов скрещивания гороха, Г. Мендель сформулировал основные закономерности наследования признаков: 1. Закон доминирования или закон единообразия гибридов первого поколения. При скрещивании особей отличающихся друг от друга по одному признаку, в первом поколении гибридов получаются потомки, схожие только с одним из родителей. Соответствующий признак другого родителя не проявляется. Проявившийся в первом поколении гибридов признак называется доминантным, а непроявившийся – рецессивным. 2. Закон расщепления гибридов 2-го поколения описывает появление во втором поколении гибридов особей с доминантными и рецессивными признаками в соотношении 3:1. 3. Закон независимого наследования признаков: при дигибридных и полигибридных скрещиваниях гибридов, каждая пара признаков наследуется независимо друг от друга и может независимо комбинироваться с другими признаками. 5. Типы наследования менделирующих признаков. 1. Аутосомно-доминантный тип наследования (Косоглазие, дальнозоркость). Критерии: · заболевание проявляется в каждом поколении без пропусков («вертикальный» тип); · каждый ребёнок больного родителя имеет 50% риск унаследовать это заболевание; · непораженные дети больных родителей свободны от мутантного гена и имеют здоровых детей; · заболевание наследуется лицами мужского и женского пола одинаково часто и со сходной клинической картиной. 2. Аутосомно-рецессивный тип наследования (Альбинизм, галактоземия - накопление в крови галактозы, которая тормозит всасывание глюкозы и оказывает токсическое действие на функцию печени, мозга, хрусталика глаза). Критерии: · заболевания с этим типом наследования проявляются только у гомозигот, которые получили по одному рецессивному гену от каждого из родителей; · родители больного ребенка, как правило, здоровы и являются гетерозиготными носителями патологического аллеля; · мальчики и девочки заболевают одинаково часто; · отмечается «горизонтальное» распределение больных, т.е. пациенты чаще встречаются в пределах одной родительской пары; · в браке двух пораженных родителей все дети будут больны. 3. Менделирующие признаки, сцепленные с полом (неполно) (болезнь Огучи – в слое палочек и колбочек, пигментом эпителии наблюдаются дегенеративные изменения; пигментная ксеродерма - заболевание, при котором под влиянием ультрафиолетовых лучей на открытых частях тела появляются пигментированные пятна. Вначале они в виде веснушек, затем в виде более крупных папиллом различной величины и, наконец, опухолей. Для большинства больных пигментная ксеродерма заканчивается летально). 6. Группы крови и резус - фактор. Группа крови — это описание индивидуальных антигенных характеристик эритроцитов. Резус-фактор – это группа антигенов, среди которых самым реакционноспособным является антиген D. По системе АВ0 группа крови может быть О (I), А (II), В (III) или АВ (IV). Группу крови определяют 2 антигена, которые расположены на эритроцитах (красных кровяных клетках) человека.
При переливании, например, эритроцитной массы, важно, чтобы она не содержала антигены, отсутствующие в крови реципиента (человека, получающего донорскую кровь)
В плазме крови человека могут содержаться антитела анти-А и анти-В (α-, β-гемагглютинины), на поверхности эритроцитов — антигены (агглютиногены) A и B, причём из белков A и анти-А содержится один и только один, то же самое — для белков B и анти-В. В случае содержания в крови (при переливании) одновременно эритроцитов с антигенами A и антител анти-A в плазме крови происходит агглютинация эритроцитов, то же происходит при наличии антигенов B и антител анти-B, на этом основана реакция агглютинации при определении группы крови системы AB0, когда берётся кровь пациента и стандартные группоспецифические сыворотки (содержащие анти-A антитела, содержащие анти-B антитела в определённом титре). В крови I группы групповые антигены A и B эритроцитов отсутствуют или их количество очень мало, поэтому раньше полагали, что кровь I группы можно переливать пациентам с другими группами в любых объёмах без опасения, так как не произойдёт агглютинации эритроцитов вливаемой крови. Однако в плазме группы I содержатся агглютинины α и β, и эту плазму можно вводить лишь в очень ограниченном объёме, при котором агглютинины донора разводятся плазмой реципиента и агглютинация эритроцитов реципиента не происходит (правило Оттенберга). В плазме IV(AB) группы агглютинины не содержатся, поэтому плазму IV(AB) группы можно переливать реципиентам любой группы (универсальное донорство плазмы). В отличие от антигенов группы крови, резус-фактор-это антиген, обнаруженный только в мембране эритроцита и не зависящий от других факторов крови. Резус-фактор передается по наследству и сохраняется в течение всей жизни человека. Если у человека отрицательный резус-фактор, то при соприкосновении с резус-положительной кровью (например, при переливании крови или во время беременности), у него могут возникнуть антитела. Эти антитела могут создать проблемы, например, во время беременности у Rh-отрицательной женщины, если у нее рождается Rh-положительный ребенок. Гемолитическая желтуха новорожденных, обусловленная иммунологическим конфликтом между матерью и плодом из-за несовместимости по эритроцитарным антигенам. Болезнь обусловлена несовместимостью плода и матери по D-резус- или АВО-антигенам, реже имеет место несовместимость по другим резус-(С, Е, с, d, e) или М-, М-, Kell-, Duffy-, Kidd-антигенам. Любой из указанных антигенов (чаще D-резус-антиген), проникая в кровь резус-отрицательной матери, вызывает образование в ее организме специфических антител. Последние через плаценту поступают в кровь плода, где разрушают соответствующие антигенсодержащие эритроциты.. Предрасполагают к развитию гемолитической болезни новорожденных нарушение проницаемости плаценты, повторные беременности и переливания крови женщине без учета резус-фактора и др. При раннем проявлении заболевания иммунологический конфликт может быть причиной преждевременных родов или выкидышей. Система Kell — это система группы крови, в которую входят 25 антигенов, в том числе самый иммуногенный после А, В и D, антиген К. На основании наличия антигена K в эритроцитах или его отсутствия все люди могут быть разделены на две группы: Kell-отрицательные и Kell-положительные. Наличие антигена К (Kell-положительный) не является патологией и передается по наследству, как и другие групповые антигены человека. Kell-отрицательным должна переливаться только кровь от доноров, не имеющих антиген К для предотвращения гемолиза. Лица же Kell-положительные являются универсальными реципиентами крови, так как у них не происходит отторжения её компонентов. 7. Хромосомы и диагностика хромосомных аномалий. Хромосомы – это структуры в ядре клетки, состоящие из ДНК и белка, которые сохраняют, реализуют и передают наследственную информацию следующему поколению. Хромосома эукариот образуется из единственной и чрезвычайно длинной молекулы ДНК, которая содержит линейную группу множества генов. Необходимыми функциональными элементами хромосомы эукариот являются центромера(1), теломеры(2) и точки инициации репликации(3). Диагностика хромосомных аномалий проводится с использованием цитогенетических методов, которые включают в себя культивирование с целью получения метафазных клеток с последующим применением дифференциального окрашивания хромосом по длине и исследованием кариотипа с помощью светового микроскопа. Данный комплекс приемов получил широкое распространение в связи с тем, что в течение долгих лет он представлял собой практически единственный способ идентификации хромосомных аномалий. Существует несколько типов дифференциального окрашивания хромосом по длине. Они определяют единую линейную дифференциацию структуры хромосом в метафазе митоза, при этом каждый метод окрашивания имеет свои характерные особенности, и существует в нескольких модификациях. Следует отметить, что они не являются альтернативными. Наиболее распространенным методом дифференциального окрашивания хромосом является G-окрашивание. Он базируется на предварительной обработке препаратов хромосом перед окраской и на использовании нефлюоресцентных красителей или их смесей. Существуют также и методы, которые окрашивают определённые участки хромосом (например, С-окрашивание, с помощью которого анализируются гетерохроматиновые участки). Применение последних также необходимо при идентификации хромосомных аномалий или дифференциации между хромосомными мутациями и морфологическими особенностями хромосом. Однако в ряде случаев применение цитогенетического анализа метафазных хромосом бывает затруднено или невозможно (сложность культивирования клеток большинства соматических тканей; диагностика хромосомных микроаберраций). 8. Классификация мутаций и их роль в эволюции. Мута́ция — стойкое (то есть такое, которое может быть унаследовано потомками данной клетки или организма) изменение генома. Классификация мутаций Первый тип классификаций По мутировавшим клеткам мутации могут быть соматические (например, разный цвет глаз у одного человека) и генеративные (или гаметические). Генеративные мутации передаются потомству, соматические проявляются у самой особи. Они передаются по наследству только при вегетативном размножении. Второй тип классификаций По исходу (значению) для организма выделяют мутации положительные, нейтральные и отрицательные. Положительные мутации появляются редко. Они повышают жизнеспособность организма и имеют значение для эволюции (например, мутации, приводящие к появлению четырехкамерного сердца в процессе эволюции хордовых). Нейтральные мутации практически не влияют на процессы жизнедеятельности (например, мутации, приводящие к наличию веснушек). Отрицательные мутации делят на полулетальные и летальные. Полулетальные мутации снижают жизнеспособность организма, сокращают срок его жизни (например, мутации, приводящие к болезни Дауна). Летальные мутации вызывают смерть организма до рождения или в момент рождения (например, мутации, приводящие к отсутствие головного мозга). Третий тип классификаций По изменению фенотипа мутации бывают морфологические (например, уменьшенные глазные яблоки, шесть пальцев на руке) и биохимические (например, альбинизм, гемофилия). Четвертый тип классификаций По изменению генотипа выделяют мутации геномные, хромосомные и генные. 1.Геномные мутации – это изменение числа хромосом под действием факторов среды. Гаплоидия – набор хромосом 1n. В природе она встречается у трутней (самцов) пчел. Жизнеспособность таких организмов снижена, так как у них проявляются все рецессивные гены. Полиплоидия – увеличение гаплоидного набора хромосом (3n, 4n, 5n). Полиплоидия используется в растениеводстве. Она приводит к повышению урожайности. Для человека гаплоидия и полиплоидия это летальные мутации. Анеуплоидия – это изменение числа хромосом в отдельных парах (2n±1, 2n±2 и так далее). Трисомия: например, если к паре половых хромосом женского организма добавляется Х-хромосома, развивается синдром трисомии Х (47, ХХХ), если она добавляется к половым хромосомам мужского организма, развивается синдром Клайнфельтера (47, ХХY). Моносомия: отсутствие одной хромосомы в паре – ♀45, Х0 – синдром Шерешевского-Тернера. Нулисомия: отсутствие пары гомологичных хромосом (для человека – летальная мутация). 2.Хромосомные мутации (или хромосомные аберрации) – это изменения структуры хромосом (межхромосомные или внутрихромосомные). Перестройки внутри одной хромосомы называются инверсии, нехватки (дефишенси и делеции), дупликации. Межхромосомные перестройки называются транслокаци.и Инверсия (отрыв участка и его поворот на 180 о ) Нехватка Делеция (выпадение среднего участка) Дефишенси (отрыв концевого участка) А B Е C D E Дупликация (удвоение участка) Транслокация (перенос участка на негомологичную хромосому) 3.Генные мутации называются точковые, или трансгенации. Они связаны с изменениями структуры генов и вызывают развитие болезней обмена веществ (их частота 2-4%). При существенном изменении условий существования те мутации, которые раньше были вредными, могут оказаться полезными. Таким образом, мутации являются материалом для естественного отбора. 9. Классификация генных заболеваний и их лечение. 1.Генные болезни с аутосомно-доминантным типом наследования. В этом случае мутантный аллель локализован на аутосоме и подавляет исходный (нормальный) аллель. Данный тип наследования встречается крайне редко. Кроме того, экспрессивность такого доминантного аллеля обычно подвержена значительным колебаниям. Это отражается в широком диапазоне клинических проявлений болезни. Если экспрессивность гена столь мала, что не проявляется у его носителя, то можно говорить о нулевой пенетрантности. Рассмотрим несколько примеров аутосомно-доминантных патологий. Ретинобластома (злокачественная опухоль глаз). Обусловлена мутантным геном хромосомы 13. Пенетрантность этого гена около 80 %. Хорея Гентингтона. Характеризуется дегенеративными изменениями мозга и прогрессирующим слабоумием. Признаки заболевания проявляются обычно после 40 лет и у гомозигот выражены значительно сильнее, что указывает на вариант неполного доминирования. Однако пенетрантность патологии высокая. Синдром Марфана. Типичны длинные тонкие конечности, высокий рост, сколиоз. Представляет собой мутацию гена, кодирующего белок фибриллин, участвующий в формировании коллагена. Характеризуется различными нарушениями скелета и связочного аппарата. Широкий диапазон экспрессивности при высокой пенетрантности. 2.Генные болезни с аутосомно-рецессивным типом наследования. Нарушения аминокислотного обмена. Общий биохимический признак - ацидоз тканей и аминоацидурия (смещение кислотно-щелочного баланса организма в сторону увеличения кислотности (уменьшению рН) и выведение повышенного количества аминокислот с мочой или наличие в моче продуктов их обмена). Неспецифические клинические признаки: рвота, обезвоживание организма (интоксикационный синдром), неврологические нарушения - летаргическое состояние или возбуждение, судорожный синдром. С возрастом появляется задержка психомоторного развития, регрессия приобретенных ранее навыков, умственная отсталость (вплоть до идиотии) и задержка физического развития. Фенотипические признаки - светлые волосы, светлая кожа, голубые глаза. Клинические симптомы ФКУ (умственная отсталость, судорожный синдром, гиперкинезы, походка, поза “портного”, склонность к дерматитам) проявляются через 3-6 месяцев после рождения. Нарушения углеводного обмена. Галактоземия. Обусловлена дефектом фермента галактозо-1-фосфат-уридил-трансферазы, что блокирует метаболизм галактозы, образующейся из дисахарида лактозы материнского молока. Симптомы заболевания (понос, рвота, желтуха) проявляются сразу после начала кормления ребенка. Смерть может наступить на первом году жизни. Частота – 1: 35 000. Аналогичные явления наблюдаются при фруктоземии, связанной с нарушением метаболизма фруктозы. Симптомы начинают проявляться после подкармливания ребенка фруктовыми соками. Обширную группу заболеваний составляют гликогенозы. Все они связаны с дефектами различных ферментов, участвующих в обмене гликогена. Прогноз многих из этих заболеваний неблагоприятный. Еще более обширную группу заболеваний образуют мукополисахаридозы – нарушения обмена гликозаминогликанов (кислых мукополисахаридов). В организме человека гликозаминогликаны, взаимодействуя с белками, образуют комплексы, являющиеся компонентами многих видов соединительной ткани. Мукополисахаридозы характеризуются значительным клиническим полиморфизмом и проявляются нарушениями функций опорно-двигательной системы, внутренних органов, умственного развития. Многие болезни имеют неблагоприятный прогноз. Нарушения липидного обмена Ганглиозидозы – накопление ганглиозида, поражающее в первую очередь клетки головного мозга. Наиболее известна из этой группы болезнь Тея – Сакса. Сфингомиелолипидозы – накопление сфингомиелина с поражением преимущественно клеток внутренних органов (болезнь Гоше, болезнь Ниманна – Пика и др.). Лейкодистрофии – нарушения метаболизма липидов, входящих в состав миелина, вследствие дефекта определенных ферментов. Известны различные клинические формы, приводящие к гибели нервных клеток и поражению головного мозга. Генные болезни, обусловленные сцепленным с полом наследованием Поскольку Y-хромосома несет незначительное число генов, в клинической генетике рассматриваются в основном мутации генов, локализованных на Х-хромосоме. Наиболее известным заболеванием этой группы является гемофилия, проявляющаяся в нарушении процесса свертывания крови. Выделяют несколько видов гемофилии (А, В) в зависимости от дефицита определенного фактора свертывания крови. Тип наследования – рецессивный, частота у мальчиков – 1: 5000. Другим сцепленным с Х-хромосомой заболеванием человека является миодистрофия Дюшенна – мутация гена белка дистрофина, участвующего в формировании мышечного волокна. Проявляется прогрессирующей слабостью скелетных мышц, нарушением работы сердца. На Х-хромосоме локализован ген фермента глюкозо-6-фосфат-дегидрогеназы. Мутация этого гена проявляется различными формами гемолитической анемии. Клиническая картина этих заболеваний включает многочисленные симптомы. Лечение Симптоматическое лечение Для ослабления симптомов наследственных болезней, связанных с дефектом определённого белка, вводят внутривенно такую его функциональную форму, которая не вызывает иммунной реакции. Такая замещающая терапия применяется при лечении гемофилии, тяжёлого комбинированного иммунодефицита и др. Иногда для компенсации определённых утраченных функций проводят трансплантацию костного мозга и других органов. Существующая терапия в подавляющем большинстве случаев мало эффективна, а само лечение следует проводить многократно, несмотря на его высокую стоимость. Так же существуют малоизученные неизлечимые болезни. Одна из них - Фибродисплазия - «Мягкая соединительная ткань, которая прогрессивно превращается в кость». Очень редкое и тяжёлое по своему течению генетическое заболевание, при котором мышцы, сухожилия и связки постепенно превращаются в кости. Процесс прогрессирует с годами, начинаясь обычно в пределах десятилетнего возраста у детей с мутацией определенного гена. Генная терапия Принципиально новым методом, эффективным и направленным на уничтожение генетической причины наследственного заболевания, является генотерапия. Суть метода генотерапии – введение нормальных генов в дефектные соматические клетки. · фетальная генотерапия, при которой чужеродную ДНК вводят в зиготу или эмбрион на ранней стадии развития; при этом ожидается, что введённый материал попадёт во все клетки реципиента (и даже в половые клетки, обеспечив тем самым передачу следующему поколению); · соматическая генотерапия, при которой генетический материал вводят только в соматические клетки, и он не передаётся половым клеткам. Риски Генотерапия может как обеспечить клиническую пользу, так и привести к расширению и злокачественной трансформации гемопоэтических клонов с переносными векторными вставками вблизи онкогенов, при использовании лентивирусных векторов, что увеличит риск лейкемии. 10. Хромосомная теория, основоположники, применение. Основные положения хромосомной теории наследственности (Т. Морган и его сотрудники). 1. Гены располагаются в хромосомах, различные хромосомы содержат неодинаковое число генов, набор генов в каждой из негомологичных хромосом уникален. 2. Гены в хромосоме расположены линейно, каждый ген занимает в хромосоме определенный локус (место). 3. Гены, расположенные в одной хромосоме, образуют группу сцепления и вместе (сцепленно) передаются потомкам, число групп сцепления равно гаплоидному набору хромосом. Наследование, сцепленное с полом — наследование какого-либо гена, находящегося в половых хромосомах. Наследование признаков, проявляющихся только у особей одного пола, но не определяемых генами, находящимися в половых хромосомах, называется наследованием, ограниченным полом. 4. Сцепление не абсолютно, т.к. в профазе мейоза может происходить кроссинговер. Дело в том, что во время мейоза при конъюгации хромосом происходит их перекрест, и гомологичные хромосомы обмениваются гомологичными участками. Это явление и есть кроссинговер (т.е. сцепленные гены могут разойтись по разным гаметам). Он может произойти в любом участке гомологичных хромосом. Сила сцепления зависит от расстояния между генами в хромосоме: чем больше расстояние, тем меньше сила сцепления, и наоборот. Расстояние между хромосомами измеряется в % кроссинговера. 1% кроссинговера, или сантиморганида, - это расстояние между двумя локусами, равная длине участка хромосомы, в пределах которого вероятность кроссинговера составляет 1%. На основе факта сцеплённого наследования Т. Морган сформулировал тезис, вошедший в генетику под названием правила Моргана: гены, локализованные в одной хромосоме, наследуются сцепленно, причём сила сцепления зависит от расстояния между генами. 11.Мультифакториальные заболевания. Мультифакториальные заболевания – это заболевания, возникающие при неблагоприятном сочетании ряда факторов: генетических особенностях (генетической предрасположенности) и влияния «внешней среды» - вредных привычек, образа жизни, профессиональной деятельности и других. В случае наследственных заболеваний мы использовали термин «мутация», а в случае мультифакториальных заболеваний – «полиморфизм» Мультифакториальные болезни можно условно разделить на: • врожденные пороки развития (расщелина губы и неба, спинно-мозговая грыжа, стеноз привратника, аненцефалия и черепно-мозговая грыжа, вывих бедра, гидроцефалия, врожденная косолапость) • распространенные психические и нервные болезни (шизофрения, эпилепсия, рассеянный склероз) • распространенные болезни «среднего» возраста (псориаз, бронхиальная астма, ревматизм, сахарный диабет, ИБС, атеросклероз, язвенная болезнь, желчекаменная и мочекаменная болезнь, ревматоидный артрит, многие формы рака). 12. Механизмы наследования групп крови и их определение у человека. От родителей ребенку передаются гены, несущие информацию о наличии или отсутствии агглютиногенов (А, В или 0), а также о наличии или отсутствии резус-фактора. Вследствие того, что наследование группы крови системы AB0 происходит по кодоминантно-рецессивному типу (2 разных доминантных гена и 1 рецессивный), фенотипические проявления происходят следующим образом: при наличии одного доминантного гена — проявляются его признаки, при наличии 2 доминантных генов — проявляются признаки обоих генов, при отсутствии доминантных генов — проявляются признаки рецессивного гена. Группа крови определяется одним геном (геном ABO), который имеет три аллели: i, IA и IB. Этот ген кодирует деятельность глюкозилтрасферазы, фермента, который изменяет углеводный состав антигенов красных кровяных клеток. Ген АВО расположен на длинном плече 9 хромосомы (9q34). По законам Менделя: · У родителей с I группой крови, будут рождаться дети, у которых отсутствуют антигены А- и В-типа. · У супругов с I и II группами – дети могут быть с I и II группами крови. · У супругов с I и III группами – дети могут быть с I и III группами крови Люди с IV группой могут иметь детей с любой группой крови, за исключением I, вне зависимости от того, антигены какого типа находятся у их партнера. Исключения возможны в крайне редких случаях, при подавлении IA и IB генов h-геном (вероятно подавление другими генами) — так называемый «бомбейский феномен». У людей, у которых данный ген находится в состоянии рецессивной гомозиготы hh, на мембране эритроцитов не синтезируются агглютиногены. Соответственно, на таких эритроцитах не образуются агглютиногены A и B, поскольку нет основы для их образования. Это приводит к тому, что носители данного типа крови являются универсальными донорами — их кровь может переливаться любому человеку, которому она нужна (естественно, с учетом резус-фактора), но в то же время, им самим может переливаться исключительно кровь людей с таким же «феноменом». Количество людей с данным фенотипом составляет примерно 0,0004 % от общей численности населения, однако в некоторых местностях, в частности, в Мумбаи (прежнее название — Бомбей), их численность составляет 0,01 %. Учитывая исключительную редкость данного типа крови, его носители вынуждены создавать собственный банк крови, поскольку в случае необходимости экстренного переливания получить необходимый материал будет практически неоткуда. Актуальность искусственной крови также сильно повышается для людей с данным фенотипом. · Более непредсказуемо наследование ребенком группы крови при союзе обладателей II и III групп. Их дети могут иметь любую из 4 групп крови с одинаковой вероятностью. Резус-фактор наследуется по аутосомно-доминантному типу наследования. Положительный резус — доминантный признак, отрицательный — рецессивный. Фенотип Rh+ проявляется как при гомозиготном, так и при гетерозиготном генотипе (++ или +–), фенотип Rh- проявляется только при гомозиготном генотипе. Если оба родителя имеют отрицательный резус-фактор, то у всех детей в их семье резус-фактор также будет отрицательный. Если один родитель имеет положительный резус-фактор, а у другого он отрицательный, то у ребенка резус-фактор может быть и тем, и другим. Если оба родителя резус-положительны, то минимум в 75% случаев ребенок также будет с положительным резус-фактором. Однако появление в такой семье малыша с отрицательным резусом не нонсенс. Это вполне вероятно, если родители гетерозиготны – т.е. имеют гены, отвечающие как за наличие положительного резус-фактора, так и отрицательного. На практике предположить это можно просто – расспросить кровных родственников. Вполне вероятно, что среди них отыщется резус-отрицательный человек. 13. Основные методы изучения наследственности в генетике. 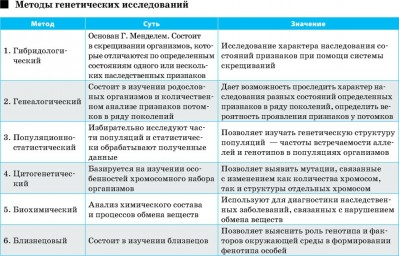 14. Аутосомы и гетеросомы. Митоз и мейоз. Митоз (непрямое деление клетки) – это наиболее часто встречающаяся форма клеточного деления, состоящая из нескольких этапов (профаза, метафаза, анафаза, телофаза). Мейоз (редукционное деление клетки) – это форма деления ядра, при котором число хромосом в клетке уменьшается вдвое, а также происходит трансформация генного аппарата. 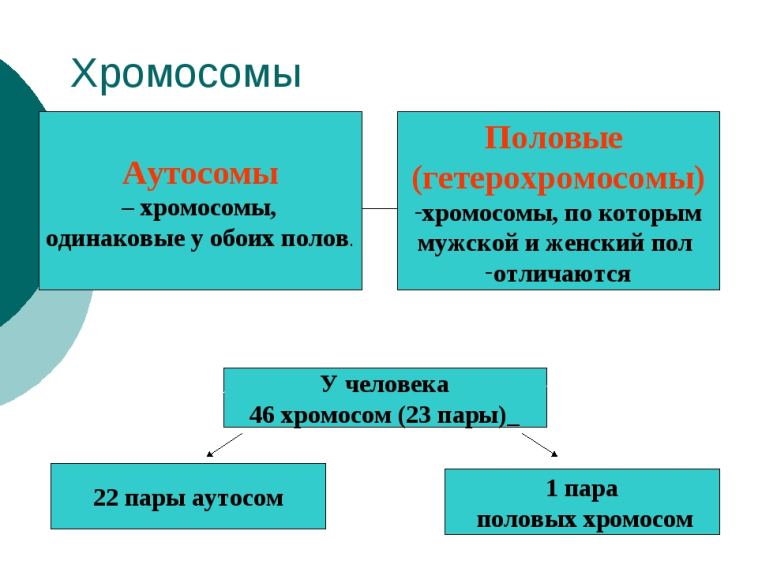 15. Медико-генетическое консультирование. Наиболее распространенным и эффективным методом профилактики наследственных болезней является медико-генетическое консультирование, которое представляет собой один из видов специализированной медицинской помощи населению, направленной на предупреждение появления в семье больного ребенка. Главная цель генетического консультирования - предупреждение рождения больных детей. Это, в первую очередь, касается тяжелых и плохо поддающихся лечению пороков развития и наследственных болезней, приводящих к физической или психической неполноценности. Задачами медико-генетического консультирования являются: 1) ретро- и проспективное консультирование семей и больных с наследственной или врожденной патологией; 2) пренатальная диагностика врожденных и наследственных заболеваний ультразвуковыми, цитогенетическими, биохимическими и молекулярно-генетическими методами; 3) помощь врачам различных специальностей в постановке диагноза аследственного или врожденного заболевания, если для этого требуются пециальные генетические методы исследования; 4) объяснение пациенту и его семье в доступной форме о величине иска иметь больное потомство и оказание им помощи в принятии решения; 5) ведение территориального регистра семей и больных с врожденной и наследственной патологией и их диспансерное наблюдение; 6) пропаганда медико-генетических знаний среди населения. Этапы: 1- это уточнение диагноза заболевания. 2- определение генетического прогноза для потомства. 3-знакомство с генетическим прогнозом для потомства. 17. Близнецовый метод исследования. Близнецовый метод основан на клиническом обследовании и сравнении моно- и дизиготных близнецов, воспитывающихся в одинаковых или различных условиях окружающей среды. Близнецовый метод дает возможность определить вклад генетических (наследственных) и средовых факторов (климат, питание, обучение, воспитание и др.) в развитии конкретных признаков или заболеваний у человека. С помощью близнецового метода удалось доказывать значение генетической предрасположенности ко многим широко распространенным заболеваниям. Результатом сравнения двух групп близнецов является расчет процента идентичности (конкордантности) различных признаков или болезней, проявляющихся у каждого из пары близнецов. Чем больше наследственная составляющая признака или заболевания, тем выше значения конкордантности, но самое главное – больше уровень расхождения между моно- и дизиготными близнецами. Количественной оценкой доли наследственной обусловленности признака является коэффициент наследуемости (Н). Если Н>70%, решающая роль в проявлении признака принадлежит наследственным факторам. При H<30% – средовые факторы являются основными в формировании признака. При промежуточных значениях Н предполагается примерно равное участие в контроле признака как генетических, так и средовых факторов. 16. Хромосомные болезни, связанные со структурой хромосом. Хромосомные болезни — наследственные заболевания, обусловленные изменением числа или структуры хромосом (геномными или хромосомными мутациями соответственно). Хромосомные болезни возникают в результате мутаций в половых клетках одного из родителей. Из поколения в поколение передаются не более 3—5 % из них. Хромосомными нарушениями обусловлены примерно 50 % спонтанных абортов и 7 % всех мертворождений. Все хромосомные болезни принято делить на две группы: аномалии числа хромосом и нарушения структуры хромосом.. Нарушения структуры хромосом Транслокации — перенос участка ДНК с одной хромосомы к другой, но не гомологичной ей. Делеции — потери участка хромосомы. Например, синдром кошачьего крика связан с делецией короткого плеча 5-й хромосомы. Признаком его служит необычный плач детей, напоминающий мяуканье или крик кошки. Это связано с патологией гортани или голосовых связок. Наиболее типичным, помимо «кошачьего крика», является умственное и физическое недоразвитие, микроцефалия (аномально уменьшенная голова). Инверсии — повороты участка хромосомы на 180 градусов. Дупликации — удвоения участка хромосомы. Изохромосомия — хромосомы с повторяющимся генетическим материалом в обоих плечах. Возникновение кольцевых хромосом — соединение двух концевых делеций в обоих плечах хромосомы. Утрата — отрыв части хромосом. В настоящее время[когда?] у человека известно более 700 заболеваний, вызванных изменением числа или структуры хромосом. Около 25 % приходится на аутосомные трисомии, 46 % — на патологию половых хромосом. Структурные перестройки составляют 10,4 %. Среди хромосомных перестроек наиболее часто встречаются транслокации и делеции 18. Хромосомные болезни, связанные с количеством хромосом. Болезни, обусловленные нарушением числа хромосом в клетках человека синдром Дауна — трисомия по 21-й хромосоме (или наличие дополнительных копий генетического материала этой хромосомы по другим причинам — за счёт транслокации или дупликации); синдром Патау — трисомия по 13-й хромосоме, характеризуется множественными пороками развития, идиотией, часто — полидактилия, нарушения строения половых органов, глухота; большинство больных не доживают до одного года; синдром Эдвардса — трисомия по 18-й хромосоме, нижняя челюсть и ротовое отверстие маленькие, глазные щели узкие и короткие, ушные раковины деформированы; 60 % детей умирают в возрасте до 3 месяцев, до года доживают лишь 10 %, основной причиной служит остановка дыхания и нарушение работы сердца. Болезни, связанные с нарушением числа половых хромосом Синдром Шерешевского — Тёрнера — отсутствие одной Х-хромосомы у женщин (45 Х0) вследствие нарушения расхождения половых хромосом; к признакам относится низкорослость, половой инфантилизм и бесплодие, различные соматические нарушения (микрогнатия, короткая шея и др.); полисомия по Х-хромосоме — включает трисомию (кариотип 47, XXX), тетрасомию (48, ХХХХ), пентасомию (49, ХХХХХ), отмечается незначительное снижение интеллекта, повышенная вероятность развития психозов и шизофрении с неблагоприятным типом течения; полисомия по Y-хромосоме — как и полисомия по X-хромосоме, включает трисомию (кариотип 47, XYY), тетрасомию (48, ХYYY), пентасомию (49, ХYYYY), клинические проявления также схожи с полисомией X-хромосомы; Синдром Клайнфельтера — полисомия по X-хромосомам у мальчиков (47, XXY), признаки: евнухоидный тип сложения, гинекомастия, слабый рост волос на лице, в подмышечных впадинах и на лобке, половой инфантилизм, бесплодие; умственное развитие отстает, однако иногда интеллект нормальный. Болезни, причиной которых является полиплоидия триплоидии, тетраплоидии и т. д.; причина — нарушение процесса мейоза вследствие мутации, в результате чего дочерняя половая клетка получает вместо гаплоидного (23) диплоидный (46) набор хромосом, то есть 69 хромосом (у мужчин кариотип 69, XYY, у женщин — 69, XXX); почти всегда летальны до рождения | ||||||||||||||||