1 Основные исторические вехи в развитии биоинформатики
 Скачать 1.58 Mb. Скачать 1.58 Mb.
|

|
6. Животные. Животный мир столь велик, что до сих пор не существует единой таксономической базы данных, охватывающей весь животный мир хотя бы до рода. Базы данных по номенклатуре и систематике созданы только для некоторых (далеко не всех) семейств, отрядов и классов животных. 9. Библиографические ресурсы в интернете и электронные публикации. Стандартные информационные поля библиографической записи в базах данных. Электрон. публикации:Многие признают, что информации как книга постепенно отмирает. Осн. препятств. в полном переходе к электрон. носителям – качествен. разница в польз-нии: книгу удобнее воспринимать. Эл. носители энергозависимы и более уязвимы, чем книга. В 2012 г. в Кодексе по номенкл-ре водорослей, грибов и раст-й было разрешено публиковать диагнозы новых таксонов только на электрон. носителе, но в «книжном» облике – PDF. Все больше стан-ся электр-х научных журналов, и такие журналы уже имеют право размещать первопубликации новых таксонов. Мн. бумажные журналы испол-т интернет как «простр-во расшир-я», публикуя там дополн. материалы к статьям, на кот. в самих статьях есть ссылки. Авторит. бумажные журналы пока держит то, что соискание ученых степеней и др.привилегии опираются на бумаж. публикации. Особен-ть поиска статьи по ключевым словам. Каждая библиографич. запись в электрон. каталоге содержит множ-во полей данных. Число полей может меняться в завис-ти от типа материала, на кот. созд-ся библиографич. запись. Для удобства поиска, а также из соображ-й совмест-ти электр-х каталогов, использ-т стандар. набор атрибутов: автор; − заглавие; − предметная рубрика; − год публикации; − место публикации; − издательство; − ключевые слова; − вид издания. Библиот. и библиограф. ресурсы в интернете: 1) Один из важнейших веб-ресурсов литературы, связанной с биотехнологией, это раздел Literature на портале NCBI .Он содержит гиперссылки на: Bookshelf – (перевод)Книжн.полка обеспеч-т свобод. доступ к книгам и документам в науке о жизни и здравоохр-нии. Полные тексты книг в формате HTML. 2) Google Books online. Полнотекст. и частично-текстовые версии книг, но все страницы – в графич. исполнении, без возможн-ти скачивания. 3) PubMed включ-т больше чем 22 миллиона цитат для биомедиц-й лит-ры от MEDLINE, журналов науки о жизни и книг онлайн. PubMed - свободная база данных, получающ. доступ к базе данных MEDLINE ссылок и резюме на науках о жизни и биомедиц-х темах. Глоб.с-ма поиска Entrez - мощная объединен. поисковая с-ма , кот. позвол-т пользователям искать данные о биотх-ии,она обеспеч-т доступ ко всем базам данных одновременно. С-ма Entrez может обеспечить представл-я о гене,последоват-х белка и хромос-х картах. Нек.учебники также доступны онлайн через с-му Entrez. 4) AGRICOLA (Сельскохозяйствен. онлайновый доступ) явл-ся базой данных, создан. и охраняемой Минист-вом сельского хоз-ва США, база данных служит каталогом и индексом для коллекций Национ.Сельскохозяйствен. Библиотеки США, но это также обеспеч-т открытый доступ инфы о сельском хоз-ве. 5) Пример электрон. тематич. библ-ки, где собраны постраничные «имиджи» книг и целых журналов: Cyberliber – An electronic library for mycology. Эта библ-ка посвящена не всей микологии, а по преимущ-ву таксономич. ее части. Она содержит: 59650 библиограф-х отчетов, 394960 просмотр-х изображ-й. 10. Геномы как объект биоинформатики. Количественные характеристики геномов C (С-value). Суммарное кол-во ДНК в гаплоидном геноме принято обозначать лат-им символом С, где "С" значит "константный" (constant) или "характерный" (characteristic), обозначая тот факт, что размер гаплоидного генома довольно постоянен внутри любого вида. Но между видами величина С широко варьирует как у прокариот, так и у эукариот. Парадокс C (C-value paradox, C-value enigma) – отсут-ие корреляции между размером генома и сложностью орга-ма. Феномен величины C: Как число пар оснований, а число генов широко варьировать от одного вида к другому, и есть только грубая корреляция между двумя (наблю-ия известный как С-значение парадокс). В насто-е время, самая высокая известное число генов составляет около 60 000, для прост-их вызывает трихомоноз.C-значение загадкой или C-значение парадокс в том, комплекс головоломка окруж-их обширную изменение размера ядра генома среди видов эукариот. В центре С-значение загадка наблюд-е, что размер генома не коррелирует с сложностью организма; Например, некоторые однокл-ые простейшие имеют геномы гораздо больше, чем у человека.Открытие некодир-ей ДНК в начале 1970-х годов решили главный вопрос C-значение парадокса: размер генома не отражает колич-во генов у эукариот, так как больш-во их ДНК не является кодир-ие и, следова-но, не состоят из генов. Геном чела, напр-р, содержит менее чем 2% белок-кодир-их областей, с остальное различные типы некод-их ДНК (особенно мобильных элементов). Базы данных по размерам геномов: Три незав-ых баз данных эукариотических инф-ии о размере генома были начаты или повторно выпущены в обнов-ой форме с 2005 года: • растительной ДНК C-значения в базе данных (www.kew.org/genomesize/homepage.html), • размер базы данных Animal Геном (WWW. genomesize.com) • размера базы данных Грибковые Геном (www.zbi.ee/fungal-genomesize/). В общей сложности, эти базы данных обесп-ют свободно доступных данных размера генома в течение> 10 000 видов эукариот, собр-ых из более чем 50 лет на сумму литер-ры. Размеры генома, как правило, дается как гамет содерж-ие ядерной ДНК («С-показатели») либо в един-х массы (пикограмм, где 1 пг = 10-12 г) или в числе пар оснований (у эукариот, чаще всего в megabases, где 1 Mb = 106 базы). Они непоср-но равноценно как 1: PG = 978 Мб. 11. Хромосомные числа и идеограммы как объект биоинформатики. «Бэндовые» карты хромосом. Указание адреса на хромосоме по международной цитогенетической номенклатуре. Хромосомное число – это условное обозн-е кол-ва хромосом в клетках и орган-х, например, 2N (ди-), 3N (три-) и 4N (тетраплоиды). Кариограмма – это изобр-е всех хромосом диплоидного набора клетки, которые распределены по группам и расположены друг за другом в порядке уменьш-я размеров с учетом индивид-х особенностей каждой хромосомы. В отличие от кариограммы, схематическое изображение хром-м наз-т идеограммой. Составление идеограммы осн-но на измерении каждой хромосомы, учете длин плеч, полож-я центромер, вторичных перетяжек и спутников, что позволят более точно идентифицировать хромосомы. «Бэндовые» карты получают путем окраски хромосом в конденсированном состоянии. Принципы условного обозначения положения полос в хромосоме таковы: для хорошо изученных организмов хромосомы пронумерованы: 1 – самая большая. Два плеча в человеческой хромосоме, разделенные центромерой, называются p (petite – короткое) и q (queue – хвостовое). Участки внутри плеча – бэнды – нумеруются 1, 2, 3… наружу от центромеры. Подразделения бэндов указывают через точку. Пример: 15q11.1. Для построения идиограмм у растений применяется компьютерный анализ изображений, в особенности для видов с мелкими и похожими хромосомами. 12. Уровни разрешения генетических карт (ГК). Принципы представления хромосомных карт в интернете. Генетическая карта— это изображ-е относит-х расстояний между генами, оцениваемых на основании измеренных частот их рекомбинации. Карты генома принято подразделять на карты генетического сцепления (собственно генетические карты) и физические карты. Различаются они методами построения. Карты генетического сцепления строятся на основе анализа данных по наследованию гена или маркера в ряду поколений, физические карты основываются на прямом исследовании носителей генетической информации – хромосом, генов, молекул ДНК. Карты генома можно строить в разном масштабе, т.е. с разным уровнем разрешения. Пример самой мелкомасштабной карты (минимум деталей) - картина дифференциального окрашивания хромосом. Максимально возможный уровень разрешения ГК – один нуклеотид. Т.е. самой крупно-масштабной картой генома явл-ся полная послед-ть нуклеотидов. Цель иссл-я геномов любых орг-в – построить крумпомасштабн. ГК. ГК высокого разрешения учитывают не только белок-кодирующие гены, но и остальные виды маркеров: *минисателлиты (участки, повтор-ся неск раз), *микросателлиты (уч-ки повторенные 10-30 раз), *ярлыки (уч-ки, состоящие из сплайсированных экзонов, локализованы в опред-ой обл-ти генома) и *перекрывающиеся послед-ти (серия перекрывающихся ДНК клонов хромосомы). Карта, построенная из перекрывающихся фрагментов, наз-ся контингом. Перекрывание устанавливается через программы выравнивания. 13.Понятие геномного проекта. Сборка генома при использовании shotgun – секвенирования: фразы, острова, скаффолды. Первый проект по геному человека завершился в июне 2000 г., и в феврале 2001 г. исследователи Международного консорциума по последовательности генома человека и компании “Celera Genomics” опубликовали два сообщения с описанием и одно – с анализом этого проекта. Однако работа по составлению полной последовательности человеческого генома продолжается до сих пор. Параллельно предпринимаются усилия по изучению полиморфизма отдельных нуклеотидов в последовательности человеческого генома и определению места, последовательности и функций всех генов. К стадии завершения подходят и многие другие программы по геномным последовательностям, в том числе для таких разных микроорганизмов. Эти геномы сейчас сравниваются с человеческим, что имеет важное значение для выявления подобия видов, возраста генов, скорости их изменения и способов функционирования уникальных генов, включая их токсичность для человека. Изучаются геномы и многих других видов, в том числе микроорганизмов, ответственных за возникновение болезненных состояний у человека. Метод дробовика (шотган-секвенирование) может быть применен для сборки сиквенсов протяженных фрагментов ДНК, в том числе хромосом и геномов. В этом слу чае ДНК случайным образом фрагментируют. Полученные мел кие сегменты секвенируют обычными методами, например, методом Сэнгера. Полученные последовательности перекры вающихся случайных фрагментов ДНК собирают с помощью специальных программ в одну большую последовательность. Существенную трудность при сборке представляют повторяю щиеся последовательности ДНК, так как они имеют участки пе рекрывания с большим числом фрагментов. Для геномов прокариот и низших эукариот метод дробовика оказался подхо дящим. Геном человека содержит огромное количество (боль ше половины) разного типа повторяющихся последовательностей. Поэтому для получения совершенной сборки необходимо применение протяженных геномных клонов. Вначале необходимо создать не сколько геномных библиотек с покрытием 5-10 эквивалентов генома. Затем клоны организуют в контиги. Контиг - это совокупность взаимно перекрывающихся геномных клонов. Скаффолд – несколько контигов, которые не удалось соединить в один, но для которых известно, что они в геноме идут подряд. Попарное определение участков пере крывания позволяет получить контиг, соответствующий доста точно протяженному участку хромосомы. Протяженные геномные клоны фрагментируют с использовани ем рестриктаз. Полученные последовательности длиной 500 700 п.н. встравают в плазмидные векторы. Эта процедура на зывается субклонированием. Затем субклонированные после довательности секвенируют и при помощи компьютерных программ собирают сиквенс всего геномного клона. 14. «Вехи» для разметки при картировании геномов: локусы рестрикции, контиги, микросателлитные локусы. Рестрикционные карты представляют собой схемы, изображающие взаимное расположение сайтов рестрикции для разных рестриктаз и расстояния между ними. Рестрикционные карты строят при исследовании протяж-х клонир-х последов-стей нуклеотидов, вкл-х исследуемые гены. РК представ-т схемы, изобр-щие взаимное распол-ние сайтов рестрикции для разных рестриктаз и расс-ния между ними. Рестрикционные карты закл-т в себе инфо об особен-х первичной стр-ры картируемых участ-в генома. Достоинство РК – инфо, кот-я в них содер-ся, м.б. испол-на непосредственно для клонир-ния интерес-х фрагм-в генома. Рез-ты анализа этих фрагм-в можно испол-ть для получения сиквенс маркирующих сайтов из данной области генома или для упорядочения ранее получ-х маркеров. Контиг (непрерывная карта клонов) - серия перекрыв-хся ДНК клонов хромосомы, хрян-хся в дрож-х или бактер-х кл-х в YACs или BACs. ДНК изуч-го орг-ма включена в маленькую доп. хромосому в дрож-й клетке, размером 1 млн. п.н. Таким образом, весь человеческий геном можно уместить в 10000 YACs. BAC может нести до 250 тыс. п.н. Однако BACs более стабильны, и работать с ними предпочтительнее. Если весь геном не секвенирован, а надо выявить местополо-е генов, то помог-т карта контигов и ярлыки. При наличии полной карты контигов для всех хромосом, то можно идентифиц-ть ярлыки, плотно сцеп-ные с интерес-щим геном, а затем локализовать эти маркеры на карте контигов. Микросателлитные последовательности ДНК встречаются с высокой частотой в геномах эукариот. Первые оценки распределения по хромосомам микросателлитных локусов были сделаны по результатам картирования генома растений и на основании прямой in situ гибридизации. При изучении распределения микросателлитов для ряда объектов, например, у сахарной свеклы, было показано, что они группируются в определенных районах хромосом. При картировании микросателлитов томатов показано, что они локализуются в прицентромерной области хромосом. Однако у большинства растительных видов микросателлитные локусы имеют более равномерное распределение по геному, что способствовало их успешному использованию в качестве маркеров как для генотипирования генома растений, так и в последующем их использовании в маркер-ориентированной селекции. 15. Аннотация генома. Основные структурные элементы, выявляемые при аннотации генома. Аннотация — процесс маркировки генов и др-х объектов в последоват-ти ДНК. Первая программная система аннотации геномов была создана еще в 1955 году Оуэном Уайтом. . Доктор Уайт построил систему для нахожд-я генов, тРНК и др объектов ДНК и сделал первые обознач-я функций этих генов. Большинство современных систем аннотации генома работают сходным образом. Аннотация генома: опис-е функциональных и структурных характеристик генома ; местонахождение кодир-х участков генов к геному, регулятор-х элементов, регулир-х транскрипцию и др-е функции генома, особенностей функционир-я генома, взаимосвязей между генами и др-х функцион-х свойств генома. Аннотация, т. е. поиск кодир-х (гены) и регулятор-х (промоторы, энхансеры) последоват-й - трудоемкий процесс, для которого можно использовать компьютерные программы. 16. Применение компьютерных программ для аннотации генома. Значение базы данных по генетическому коду. Программы, использ-е при аннотации генома: 1. GENEMARK - Программа предсказания семейств генов. Позволяет предсказ-ть потенциальные белок-кодирующие участки в геномах про-,эукариот, вирусов и плазмид. 2. GeneFinder - Программа поиска сайтов сплайсинга, кодирующих экзонов, промотров, поли-A сигналов. 3. GeneScan - Программа предсказания экзонов, интронов, и границ между ними. 4. Прочие программы для аннотации геномов растений: Поиск экзонов в геноме арабидопсиса - Arabidopsis Internal Coding Exon Finder - Программа позволяет вставить ваш сикенс в спец-е окошко и получить ответ о найденных (или не найденных) экзонах на ваш e-mail адрес. Arabidopsis GenSequer - Программа, предсказ-я точки сплайсинга в геноме арабидопсиса. TSSP-TCM - Программа определ-я промоторов в геномах растений. 5. Программа поиска повторов – RepeatMasker. Позволяет отправить ваш сиквенс онлайн для обработки программой, и получить ответ о наличие повторов и модифицир-й сиквенс, в котором повторы будут отмечены. Программа ищет: a) собственно рассеянные повторы, б) участки ДНК упрощенной струк-ры 6. MicroScope - Microbial Genome Annotation & Analysis Platform. Интернет-платформа для сравнительного анализа и функцион-й и структур-й аннотации микробных геномов. База данных по генетич-му коду различных организмов - Codon Usage Database. В ней представлены статистич-е таблицы кодонов. 17. Процедура выявления (локализации) генов в геноме на основе сиквенсов. EST-последовательности. Базы данных по промоторам. В настоящее время существует довольно большое количество компьютерных программ для выявления начала гена в известных сиквенсах. Программа определяет открытые рамки считывания (ORF) ― районы ДНК, которые начинаются со старт-кодона (ATG; у прокариот также GTG и CTG) и заканчиваются стоп-кодонами (TAA, TAG, TGA в стандартном генетическом коде). Таким образом, ORF ― это потенциальный белок-кодирующий фрагмент. Выявление белок-кодирующих областей ведется 2 путями: (1) Определение сиквенсов, похожих на известные белок-кодирующие области у других организмов. Они могут быть похожи либо на (а) известные белки, (б) известные EST ― экспрессируемые последовательности. Первый подход основан на определении районов похожих на известные белок-кодирующие области из других организмов. Эти районы могут кодировать аминокислотные последовательности, похожие на известные белки, или могут быть похожими на ярлыки экспрессируемых последовательностей (EST, expressed sequence tag). Поскольку EST опре-делены из мРНК, они соответствуют генам, о которых точно известно, что они экспрессируются. В этом случае необходимо секвенировать всего несколько сотен начальных нуклеотидов кДНК, чтобы получить достаточно информации для идентификации гена. (2) Поиск и идентификация генов ab initio (с начала), на основе только сиквенса. Эта процедура значительно более проста для прокариотических геномов, т.к. гены прокариот непрерывны, а межгенные промежутки довольно малы. Ярлык экспрессируемой последовательности EST (Expressed Sequence Tags) – это секвенированный отрезок последовательности клона, случайно отобранного из библиотеки кДНК, используемый для опознавания генов, экспрессируемых в определённой ткани. Мы далеко не всегда располагаем расшифровками полных последовательностей ДНК; в основном накопленные к настоящему времени данные о ДНК состоят из отдельных отрезков последовательностей, большая часть которых представлена ярлыками экспрессируемых последовательностей (EST). В анализе EST-последовательностей необходимо учитывать следующие моменты: 1) алфавит EST-последовательностей состоит из пяти знаков (a, c, g, t, u), 2) в последовательности могут присутствовать фантомные всуды (сокр. вставка/удаление – insdel, от англ. insertion/deletion – инсерция/делеция), приводящие к сдвигам рамки трансляции; 3) весьма вероятно, что EST-последовательность окажется подпоследовательностью какой-либо последовательности из баз данных; 4) EST-последовательность может вовсе не представлять отрезок кодирующей последовательности (coding sequence) какого-либо гена. Принцип секвенирования EST-последовательностей показан на рисунке 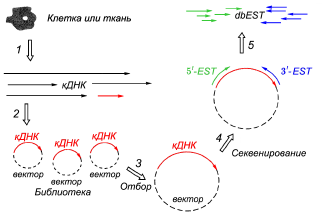
В GenBank коллекция dbEST на настоящий момент содержит почти 9*106 статей для 348 видов. |