1 Основные исторические вехи в развитии биоинформатики
 Скачать 1.58 Mb. Скачать 1.58 Mb.
|

|
25. Поиск последовательностей в GenBank: инструмент BLAST и его значение. Использование количества очков (score) и величины Е при поиске. Источники информационного «шума» в генетическом банке. Чтобы данные, хранящиеся в GenBank, находились в свободном on-line доступе и были связаны с публикациями, админ-ция генетич. банка требует цитировать идентификац. номера сиквенсов в научных статьях, и журналы требуют внесения новых сиквенсов в GenBank и получ. соотвеств-х учетных номеров.Разраб-но большое кол-во программ для сравн. последоват-тей с последующ. определен.их сходства, но часто использ-ся программы серии BLAST.Сюда входят программы для нахожд. лок-го выравнив. между заданной последоват-тью и последоват-тями из базы данных.Сем-во программ серии BLAST делят на 7 групп:1.Геномные программы–для сравн. изучаемой нуклеот. последоват-ти с базой данных секвениров-го генома.2.Нуклеотидные–для сравн. изучаемой нуклеот-й последоват-ти с базой данных секвениров-х нуклеин-х к-т и их участков. 3. Белковые–для сравн. изучаемой аминокисл-й последоват-ти белка с имеющейся базой данных белков и их участков. Туда входят алгоритмы4. blastx–сравнивает транслирован.послед-ть с базой данных белковых последоват-тей.5. tblastn–сравнивает аминокислот. послед-ть с базой данных транслирован.нуклеотидн. последоват-й.6.tblastx–сравнивает транслирован. последоват-ь ДНК с базой данных транслиров-х нуклеот-х последоват-тей.7. Специализированный • cdart–сравн. с целью поиска гомолог-х белков по доменной архитектуре;• VecScreen – определ.сегментов нуклеот-й последоват-ти нуклеиновой к-ты, кот. могут иметь векторное происхожд-е.•bl2seq–локал. выравн-е двух последоват-тей и др. Принцип работы BLAST.Сначала алгоритм BLAST созд-т таблицу всех «близких» слов фиксирован. длины(по умолчанию–длины 3 для белк-х последоват-тей, 11 — для нуклеот-х),кот. бы локально выравнивались с заданной последоват-тью.Вес выравн-я д.б. выше порогового знач-я. Затем алгоритм сканирует базу данных, и когда находит слово из списка, нач-т процесс «расшир-я совпадения»,чтобы увеличить возможный участок выравнив-я без разрывов, в обоих направл-х, до достиж. макс.веса. После вычисл-ся статистич. значимость найденных совпад-й, и если она превышает определ. порог, то выдается резул-т. Результат поиска, например в blastn, включ-т в себя:1) Графич. изображ-е обнаруж-х гомологов;2) Список гомологов с оценкой значим-ти находки;Для каждой обнаружен. последоват-ти необх-мо опред-ть значимость сходства с изучаемой последоват-тью. Для этого программа вычисляет вес (score) выравнивания и величину E. Вел-на E показывает значимость найден. совпадения, т.е. показывает, насколько случ-м м.б. найден. совпадение.Чем выше вес,тем больше сходство двух последоват-тей. Чем меньше величина Е, тем достовернее выравнив-е.Гомология ниже 50 % при больших значениях вел-ны E несущественна.3) Локал. парные выравнив-я нуклеот-й последоват-ти с последоват-ми гомологов.Т.о., BLAST вычисляет для каждого найденного сиквенса:а) колич-во очков согласно сопадениям;б) вел-ну E. Источ-ки «информац-го шума» в генетич. банке.1) Сиквенсы, принадлежащ. к загрязняющему, а не целевому огр-му.2) Химерные вставки. 26. Понятие выравнивания последовательностей нуклеиновых кислот и его биологическое толкование. Элемент-ые явления в про-цессе эволюции последовательностей. Вырав-е последов-ей – это опред-ие соответствий между остатками (или основаниями). Цель выравн-ия – устан-ие вероятных нуклео-ых замен, вставок и делеций, кот-е отличают между собой родственные (гомол-ые) сиквенсы, и которые могли происходить в ходе эволюции последов-тей. Количество мутаций (различий) между последов-ми указывает на степень их расхож-ия в ходе эволюции. Если сиквенсы неродственны (негомологичны), то вырав-ние будет слишком спекулятивным, и лишенным биолог-го смысла. Цели сравнения двух послед-тей состоят в: (1) измерении сходства почленно; (2) нахождении консерват-ых и вариабельных областей; (3) высказвании предпол-ий об эволюц-ной связи между последов-ями. Выравнивание прим-тся для (1) построении эволюц-ых деревьев; (2) аннотации геномов, опред-ии структуры и функции генов. Выравн-ие кодирующих последоват-ей НК в биолог-ом смысле тесно связано с выравн-ем производных от них аминок-ых последов-ей. Сравнение двух предпо-но гомологичных последов-ей показывает степень их рас-ия, то есть силу эволюционных изменений. Вырав-ние последовате-тей – это процедура сравнения двух (попарное выравнивание) или нескольких (множественное выравнивание)последов-стей путём поиска рядов отдельных элементов или хара-ых комбинаций элементов последов-тей, кот-е распо-ены в выравниваемых последов-тях в одинаковом порядке. При вырав-ии двух последов-стей их помещают в две строки друг над другом, записывая их с помощью букв алфавита. Идентичные или подобные "буквы" (элементы) этих строк (посл-стей) сдвигают в пределах строки (не меняя исходного порядка следования "знаков") т. о., чтобы они выстраивались друг под другом в соотве-щих столбцах. Неидентичные, или различные знаки либо помещают в одни и те же столбцы как несовпадения, либо вставляют напротив них во второй последо-сти пропуски. типы выравнивания. Глобальное вырав-ие –это вырав-ие всей последо-ти относительно другой последо-сти. Локальное выравн-ие –это поиск части последо-сти,которая совпадает с частью другой послед-ти. Поиск мотивов совпадения – это поиск совпадения короткой посл-сти в одном или более отрезках длинной послед-сти. В этом случае допускается несовпадение одного символа. Можно так-же потребовать полного совпадения, либо допустить бόльшее число несовпадений или даже пропусков. Множественное выравнивание –это взаимное выра-ние многихпоследова-тей. 27. Парное и множественное, глобальное и локальное выравн-ние. Основные проблемы при выравн-ии последовательностей (…). Выравн-е послед-тей —биоинформатич-ий метод, осн-ный на размещении двух или более послед-тей мономеров ДНК, РНК или белков др. под другом т.о., чтобы легко увидеть сходные участки в этих последов-х. Осущ-ся с учетом как замен, так и вставок/делеций. Если выравн-ся 2 послед-сти, то такое выравнивание называется парным (рair sequence alignment). А если проводят выравн-е 3 или более послед-тей одновр-но – множественным (multiple sequence alignment). Для такого выравн-я созданы специальные прогр-мы: CLUSTALW, MAP (мы ей пользовались на парах) и Pima. Различают полное и частичное выравн-е. Полное или глобальное выравнивание (global alignment) – это выравнивание нуклеотидных или белковых последовательностей по их полной длине. Gример, для двух аминокисл-х послед-тей A. Частичное или локальное выравнивание (local alignment) – выравн-е части нуклеот-х или белковых последов-тей. Для вышеприведенных послед-тей пример. А HEAGAWGHE –E – –-– AW– HEAE В ….AWGHE…. ….AW– HE… Используются разные алгоритмы и методы выравнивания. Эмпирическим показателем количественной оценки качества выравнивания является его вес (score). Чем выше вес, тем больше сходство между последовательностями и выше качество выравнивания. Оптимальное выравнивание (optimal alignment) – это выравнивание нуклеотидных или белковых последовательностей с самым высоким весом и имеющее биологический смысл. Вес выравнивания рассчитывается исходя из количества замен, с учетом разрывов и т.н. матрицы замен. Главнейшие проблемы при выравнивании: а) проблема инделей: что исследуемый геном отличается от уже изв-го из-за инделов (мутации по инсерции (вставки) либо делеции нуклеотидов). Из-за этого последующей цепочке будет не совпадать, поэтому программы выравнивания должны уметь находить точечные несоответствия и правильно соотносить следующие. б) проблема концевых разрывов … VHLTPEEKSAVTALWGKVNVDEVGGEAL NEEDVGGEALSAVHLVGTSA… Заключается в том, что когда мы берем для выравнивания две последовательности аминокислот С-участок первой посл-ти (ее конец) начинает совпадать, т.е. выравниваться с N-концевым участком (началом) второй последовательности, т.е. это выравнивание получится почти бессмысленным. в) При наличии тандемных повторов не всегда можно однозначно решить, как нужно выравнивать две последовательности: либо дальнейшем наложением, либо с обособлением данного повтора (условным вырезанием при наложении). Такую последовательность можно картировать случайным образом на любой подходящий участок или же пометить как картирующуюся на множественные участки. AGTCGACGACGAGCGATCTGGAT AGTCGAGCTAACTGGAT AGTCGAGCGATCTGGAT AGTCGAGCTAACTGGAT г) иногда в геноме организмов встречают боксы полной неопределенности, ничего не кодирующие и не выполняющие никаких функций. При выравнивании эти участки сравнивать друг/с другом не следует, т.к. это снизит общий процент совпадений, хотя боксы роли совершенно не несут. д) высоко вариабельные домены часто встречаются в белках, с некоторой стороны они похожи на боксы полной неопределенности, в выравнив-ии учитываются, но значимости не имеют. 28.Точечные матрицы сходства. Меры сходства последовательносетй и показатели качества выравнивания. Штрафы за разрывы. Точечные матрицы сходства –это простейшие изображения, дающие представления о сходстве между двумя последовательностями. Матрица сходства логически связана с выравниваниями. Строки и колонки матрицы отвечают основаниям в первом и втором сиквенсе. Ячейки матрицы оставляют пустыми, если основания различны, и заполняют, если сходны. Совпадающие фрагменты в конечном итоге отразятся в виде диагоналей, идущих от левого верхнего угла в правый нижний. 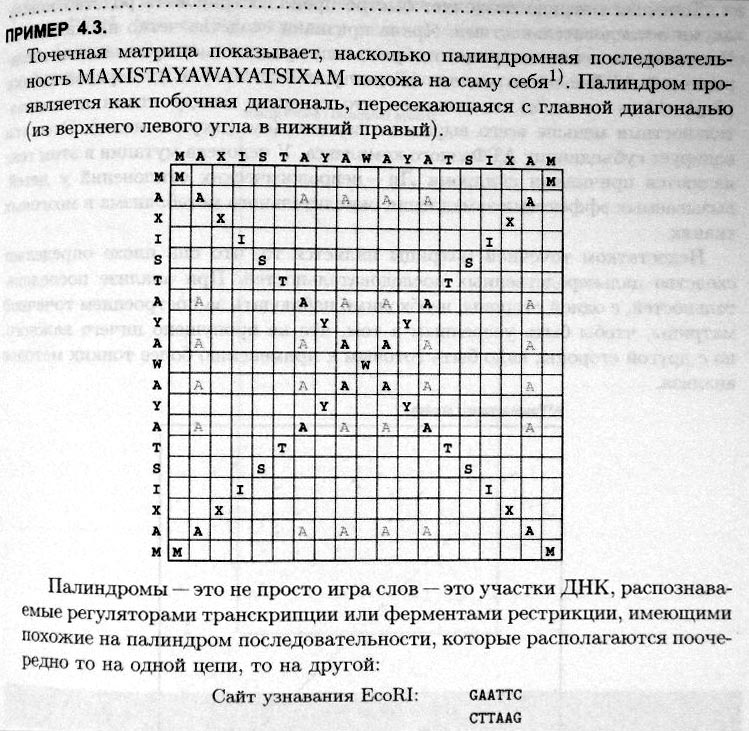 Биологическое значение палиндромов: (1) распознаются регуляторами транскрипции (2) узнаются эндонуклеазами (3)являются участком связывания белка-димера, плечи которого взаимодействует с плечами палиндрома. Точечная матрица объединяет в одном изображении не только полную информацию о сходстве двух последовательностей, но также представляет полный набор и относительное качество различных вариантов выравнивания. Любой путь из левого верхнего угла к правому нижнему, двигаясь при каждом шаге только строго направо, вниз и по диагонали, соответствует возможному выравниванию. Если последовательности близкородственны, то выравнивание может быть считано непосредственно с графика. Ни одно движение не может совершаться вверх и влево, так как при этом один нуклеотид последовательности 1 будет сравниваться сразу с несколькими в последовательности 2. Горизонтальное передвижение указывает на вставку, вертикальное – на пропуск (делецию). Расположение диагонали сходства в точечной матрице сходства указывает на молекулярное родство последовательностей. Например, диагональ может быть сплошной только в конце или только в начале сравниваемых последовательностей. Меры сходства последовательностей. Меры сходства указывают на подобие последовательностей, после того, как произведено выравнивание. Существует 2 метода измерения сходства между сиквенсами:  Для НК транзиции [пурин <-> пурин] и [пиримидин <-> пиримидин] (a <-> g, t <-> c) происходят чаще трансверзий [пурин <-> пиримидин] [(a, g) <-> (t, c)]. Кроме того, делеция группы расположенных рядом нуклеотидов гораздо более вероятна, чем делеция нуклеотидов, не расположенных рядом. По этой причине при поиске оптимального выравнивания компьютерная программа может назначать «цены» за каждый из видов замен либо инделов. Складывая цены, программа может присвоить общую цену для каждого из вариантов выравнивания. Расстояние по Хэммингу и Левенштайну указывают на степень несходства между сиквенсами. В то же время мера сходства опирается на суммарные очки, присвоенные всем совпадениям между сиквенсами. Штрафы за разрывы. Разрыв (gap) – прочерк (–), который вводят в выравнивание для компенсации вставки (выпадения) нуклеотидов или аминокислот в одной последовательности относительно другой. Для предотвращения накопления слишком большого числа разрывов в выравнивании при введении очередного разрыва из общего веса выравнивания вычитается установленный штраф. Дополнительный штраф может применяться для контроля длины разрыва, то есть числа подряд идущих пробелов. Самый простой вид штрафа – так называемый линейный штраф, пропорциональный длине разрыва: R(g)= – gd ,где g– длина разрыва; d – штраф за одиночный разрыв. Другой вид, так называемый аффинный штраф за разрыв определяют по формуле: R(g)= – d – (g – 1)e, где g– длина разрыва;d – штраф за открытие разрыва; е – штраф за его продолжение. Обычно штраф за продолжение разрыва (е) меньше штрафа за открытие (d);тогда длинные вставки и делеции аффинной функцией штрафа наказываются меньше, чем линейной. Это желательно, когда известно заранее, что ожидаемая частота разрывов в один и несколько остатков примерно одинакова. Типичные значения штрафов за разрывы, используемые на практике, равны d= 8 для линейного штрафа, или d= 12, e= 2 для аффинного случая. 29.Компьютерные программы выравнивания .Результаты множественного выравнивания (МВ): мотивы и блоки. Однонуклеотидный полиморфизм как источник молекулярно-биологической информации Выравнивание – это сопостав-ление 2х и более последов-ей для опреде-ия их уровня идентичности с учетом замен и вставок/делеций. Компьютерные программы по выравниванию. По механизму исп-ния они дел-ся на 2 группы: серверные – кот-е решают задачи, передаваемые с комп-ра пользователя на сервер, и загружаемые – решают задачи, будучи установ-ми на компьютер пользователя. 1. ClustalW (download) 2. BioEdit (download) 3. MAFFT (server) 4. MUSCLE (server) 5. T-Coffee (server & download) Цель использ-ния программ по выравниванию на примере программ серии CLUSTAL. Основн.предназн-ем выравн-й пом-ю программ Clustal, является вычисл-ние на их основании т.н. эволюц-ых расст-й между аминокис-ми или нуклеот-ми последовательностями, определ-е типа аминокисл-х замен, поиск функцион-но важных участков. В ходе выравн-ния выявл-ся консервативные участки последоват-тей, кот-е могут явл-ся элементами вторичной стр-ры, сайтами связывания лигандов. Прогр-мы семей-ва Clustal исп-ся для построения дендрограмм, показыв-х филогенет-кие отнош-ия сравнив-х послед-стей. Множественное выравнивание- это вырав-ние набора из 3х и более последова-стей одновременно, при кот-м элементы в одинак-х позициях группируются в колонки. Множ-ое выравнивание выявляет высоко консервативные участки («блоки»), сходные у всех сиквенсов, менее консервативные позиции, в которых у отдельных сиквенсов имеются нуклеотидные замены, и вариабельные участки, изобилующие заменами и инделами. Однонуклеотидный полиморфизм(ОНП)- это генетич.изменчивость между особями.Условия возникновения ограничены одной начал-ой парой, в кот-ой может возн-тьзамена, вставка или делеция. Злокачественная анемия,пример заболевания,вызв-го специфичным ОНП: замена А на Т в бета-глобулиновом гене вызывает замену Глу на Вал, делая поверхность гемоглобина способной к слипанию,что ведет к полимеризации бескислородной дезокси-формы.ОНП,рассредоточенные по всему геному встр-ся в среднем один раз на 2000 пар.Немотря на точ,что они выз-ся мутациями, многие позиции ,сод-щие ОНП,имеют низкие уровни мутации м.б. исп-ны в кач-ве стаб-ыхмаркеров для картирования генов.Не все ОНП связаны с заболеванием.Многие из них встр-ся внутри нефункциональных областей. Нек-ые ОНП происх-т внутри экзонов,вызывая замену на синонимичный кодон или замену, незначительно влияющую на функциональность белка.Другие типы ОНП могут выз-ть измен-я в белке до сер-го уровня ,чем локальные: 1)замена значащего кодона на стоп-кодон,что приводит к преждевременной остановке синтеза белка,2) делеция или вставка приводит к сдвигу рамки считывания.зависимость между забол-ем и специф.СОНП м.б. исп-на в леч.практике, так как это легко обнар-ть у больных. Но если заболевание проис-т из-за дисфункции специфичного белка,то м.б. много сайтов мутации,кот-е могут вызвать эту иннактивацию.Конкретный сайт может доминировать,если носители гена явл-ся потомками одной особи,у кот-й произошла мутация или если забол-ие возникает в ре-те получения какй-либо специф-ой особенности (пример серповидноклеточная анемия: способность гемоглобина серповидных кл-е полимеризоваться) 30. Зависимость выравнивания рибонуклеиновых кислот от вторичной и третичной структуры РНК. Выровненные сем-ва генов - Важнейшие веб-ресурсы: 1. HOGENOM: Database of Complete Genome Homologous Genes Families. (производная от базы данных по гомологичным генам бактерий). 2. HOVERGEN : Homologous Vertebrate Genes Database. Завис-ть выравн-я от втор-й и третичной стр-ры. Такая завис-ть среди НК сущ-т для молекул РНК, которые имеют специфич-е вторичную и третичную стр-ру. Структуры РНК: первичная (линейная) – это сиквенс мономерных единиц, объед-х ковал-ми связями; вторичная – обобщенная 3-х-мерная форма локальных сегм-в цепи РНК, опред-я водор-ми связями; третичная – специф-е положение отд-х атомов в трехмерной структуре молекулы, т.е. описание молекулы через атомные координаты (отличие от вторичной – более точное описание 3-х-мерной структуры). Осн-е эл-ты 2-чной стуктуры РНК: петля (шпилька), участок двойной цепи, двустороннее вздутие, одностороннее вздутие. Осн-е эл-ты третичной структуры РНК: всевдоузелок, целующиеся шпильки, контакт шпильки и вздутия. Осн-я рабочая гипотеза при предсказ-и стр-ры РНК: Естеств-я вторичная стр-ра РНК – это укладка молекулы т.о., чтобы она имела наименьшую свободную энергию. Свободная энергия уменьш-ся за счет образ-я водородных связей. Структ-е выравн-я, кот-е обычно специфичны для белков и иногда послед-й РНК, исп-т инф-ю о вторичной и третичной стр-ры белка или РНК молекулы, чтобы помочь в выравн-и послед-й. Эти методы могут быть использованы для двух или более послед-й. Структ-е выравн-я исп-ся в качестве "золотого стандарта" в оценке выравн-я для гомологии на основе предсказания стр-ры белка, потому что они могут выровнять участки белковой послед-ти, кот-е структурно похожи. Принципы структурного выравнивания и предсказания структуры РНК: 1. Мол-лы со сходными фу-ми, но с разными сиквенсами, имеют тенденцию формир-ть сходные структуры; 2. инвертированные уч-ки одинаковой длины, с взаимно комплиментарными сиквенсами, указ-т на формир-е уч-в комплиментарного спаривания; 3. участки сиквенсов между инвертированными уч-ми указывают на формирование петель; 4. наличие ковариабельных позиций (изм-е в одной позиции влечен изм-е в другой, удаленной от нее) указ-т на сайты гибридизации; |