1 Основные исторические вехи в развитии биоинформатики
 Скачать 1.58 Mb. Скачать 1.58 Mb.
|

|
3D-профиль. Авторы определяют 6 классов АК на основе доступности и полярности окружения. Боковые цепи каждого из классов могут быть в любом из 3 типов вторичной структуры. Т.о. всего получается 18 классов. Если отнести каждую боковую цепь к одному из 18 классов, то можно, пользуясь алфавитом из 18 букв, создать описание белковой структуры, называемой профилем трехмерной структуры (3D- профилем). Можно выравн-ть 2 далекие друг от друга родственные послед-ти путем выравн-я их 3D- профилей, а не самих АК. Метод 3D- профилей превр-т белковые стр-ры в одномерные объекты, не сохран-щие точно ни послед-ть, ни стр-ру мол-л. Преимущество метода в том, что он может быть автоматизирован. Новую послед-сть можно сравнивать с каждым 3D- профилем в библиотеке известных фолдов. Основные типы графического предст-я белковых структур (типы, встр-ся в трех основных видах визуалзаторов: PV, Jmo, JSmol – посл. мы пользовались на парах):* Backbone дает 'схематичное' представление белка в виде изогутых разноцветных линий, каждая из кот представл собой отдельный домен / функциональный участок (потребляет меньше всего графических ресурсов , в отличии от других видов). * Molecular Surface=Spase Fill наиб-е полно отражает именно упаковку атомов в простр-ве белк-й стр-ры. Каждый атом наглядно представлен в виде сферы с радиусом Ван-дер-Ваальса. Но здесь не видны связи цепочек АК. *Sticks - палки - дает представление именно о связях АК в цепочке - здесь они четко видны (но не видны отдельные атомы.) Удобно смотреть белок именно как "цепь аминокислот". *Ball & stick - шарики и палки - такой же как и третий, но здесь в дополнении к связям видны еще и атомы. *Cartoon – видны вторичные спирали, бета-слои и связующие их цепочки АК. Кроме того, во всех режимах можно вкл-ть режим подписи каждой АК. 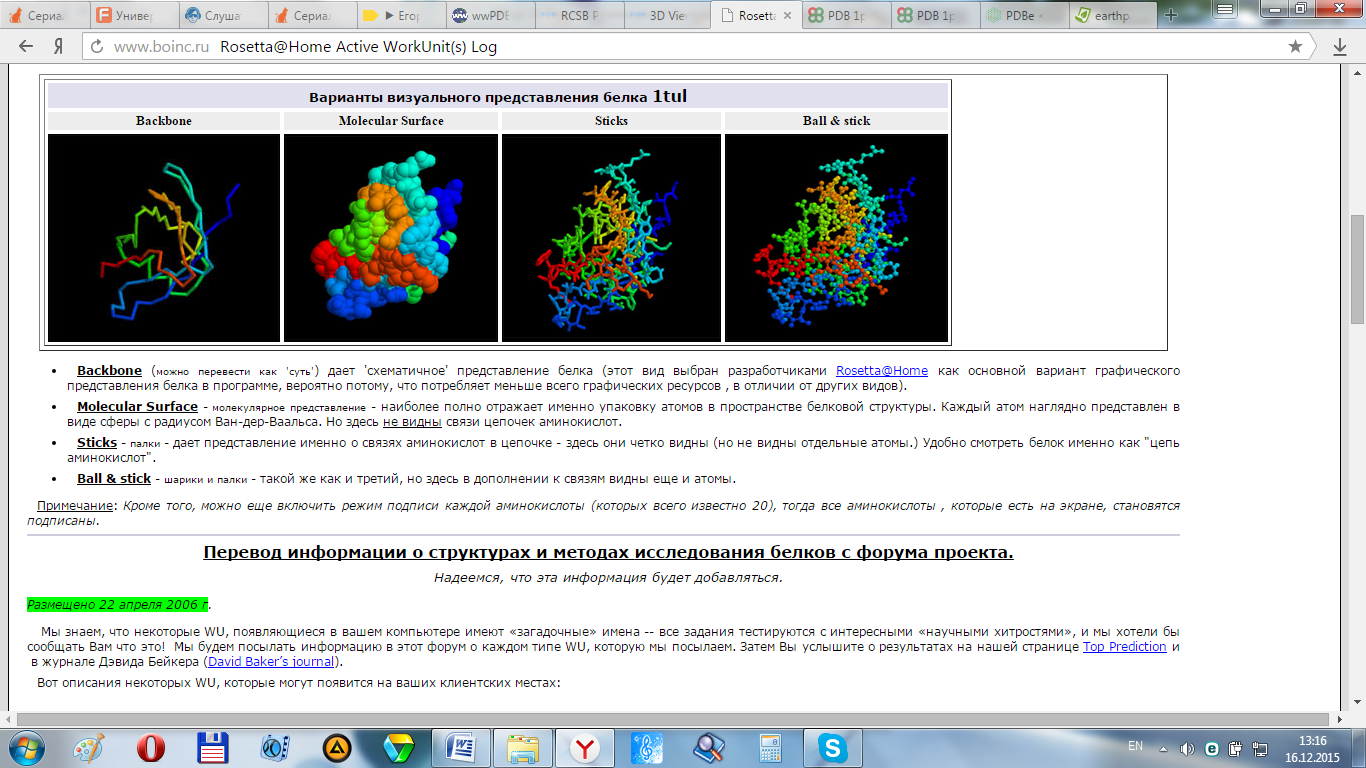 44.Принципы сворачивания(фолдинга) белков. Роль физико-химических характеристик боковых остатков в формировании белковой стр-ры. Гидрофобный эффект. В ходе нескольких независимых экспериментов по рефолдингу в определенный момент времени образец подвергается обработке протонами. После того так нативная конформация образовалась, определяется, где произошел изотопный обмен. Эти исследования подтверждают механизм сворачивания через образование «расплавленной глобулы», содержащей правильные участки вторичной структуры, которые не стабилизируются взаимодействиями, присущими третичной структуре. В дальнейшем происходит сворачивание «расплавленной глобулы» в супервторичную структуру, постепенно приближаясь к конечной, нативной конформации. Именно боковые остатки а/к создают то разнообразие физико-химических свойств, которое порождает различные способы укладки. Боковые радикалы 20 а/к различаются по следующим параметрам: Размер. Самая маленькая а/к глицин содержит в боковой цепи только 1 атом водорода, а одна из самых больших фенилаланин- бинзильный радикал. Электрический заряд. Некоторые боковые остатки несут + или – заряд при нейтральном pH. Разноименно заряженные остатки могут попарно взаимодействовать, образовывая солевые мостики. Полярность. Некоторые остатки полярны; они могут образовывать водородные связи как с другими полярными остатками, так и с основной цепью или с водой. Взаимодействие углеводородов с водой термодинамически невыгодно, такие остатки называют гидрофобными. Слипание гидрофобных остатков во внутренней области белков вносит существенный вклад в стабилизацию структуры белка. Форма и жесткость. Конформация боковой цепи зависит от ее химической структуры и от ее внутренней конформационной подвижности. Гидрофобный эффект: Неполярные боковые цепи белков ведут себя в растворе как масло. Их взаимодействие с водой невыгодны. Кузман, предположил, что они будут сближены внутри белка, изолированно от растворителя, подобно молекулам в капле масла. Эта модель капли масла была подтверждена с помощью структур глобулярных белков. Как следствие гидрофобного эффекта, заряженные остатки практически исключаются из внутренностей белковой глобулы; в редких случаях они формируют внутренние солевые мостики. Остов должен проходить внутри белковой структуры и нести с собой полярные атомы N и O, способные взаимодействовать с другими полярными атомами главной цепи и полярными боковыми цепями. 45.Предсказание стр-ры белка. Типы задач по предсказанию стр-ры белка Предсказание пространст-й стр-ры белка по аминокисл-й последов-сти явл-ся одной из важных задач биоинформатики. Простран-ная стр-ра белка тесно связана с его функционированием. Число известных перв-х белк-х стр-р, определ-х по нуклеотидным последоват-ям ДНК, превосходит число известных пространств-ых белк-х стр-р. Зная перв-ую стр-ру белка и его предсказанную 3мерную пространс-ную структуру можно предсказать функцию белка. Есть м-ды для предсказания пространс-ной стр-ры белка по его аминоки-ной последов-сти. Широко приме-ый метод основан на исп-нии инфо из баз данных 3хмерных структур белков.В этом методе исп-т инфо об известной пространст-й стр-ре белков, облад-х первичной структурой близкой к исследуемому белку. Одной из программ, с пом-ю к-й можно предсказать 3чную стр-ру белка, принимая за основу уже известную 3чную стр-ру ближайшего гомолога - Geno3D. SAM-Т99. В этой программе вначале произв-ся поиск в базе данных аминокисл-х последоват-тей, гомолог-х заданной, и по ним настраивается профиль – основа скрытой марковской модели. Получ-ая модель далее испол-ся для поиска белков, слабо гомолог-х заданным аминокисл-м последоват-тям. PSIPRED позволяет предсказывать простран-ную стр-ру белка по аминокис-ой последоват-сти 3 мет-ми: 1) PSIPRED –распозн-ние вторичной стр-ры, осн-ное на нейронных сетях; 2) MEMSTAT – предсказание втор-й стр-ры и топологии трансмем-ных белков, испол-щее множес-ные выравнивания, пол-ные из PSI-BLAST; 3) GenTHREADED – распознавание 2чной стр-ры и поиск родст-ных последо-стей, использующее алгоритм выравнивания по профилю. Базы данных простран-х структур: PDB (Brookhaven Protein DataBank) – коллекция 3D–структур биолог-х макромолекул экспериментально определ-х с пом-ю рентгенострук-го м-да. PFAM (Protein families database of alignments and HMMs) – большая коллекция белковых семейств. InterPro – база данных белковых семейств, доменов и функц-х сайтов, найденные в известных белках. 46. Особенностити выравн-я аминокисл-х послед-й. Примен-е BLAST для поиска сходных аминокисл-х послед-й. Основные веб-ресурсы по протеомике. Выравн-е – это сопоставл-е двух и более послед-й для определения их уровня идентичности с учетом как замен, так и вставок. Это способ написать послед-ти друг под другом так, чтобы гомологичные буквенные обозн-я стояли друг под другом. Если выравн-ся две послед-ти, то такое выравн-е наз-ся парным. А если проводят выравн-е 3 или более послед-й одновр-но – множественным. Разл-т полное и частичное выравн-е. Полное или глобальное выравн-е – это выравн-е нуклеот-х или белковых послед-й по их полной длине. Частичное или локальное выравн-е – выравн-е части нуклеот-х или белковых послед-й. Важным звеном исслед-й молек-й биологии явл-ся сравн-е аминокисл-х и нуклеот-х послед-й, кот-е позв-т идентиф-ть сем-ва генов, относить к ним секвенир-е послед-ти, устан-ть их структ-е и функц-е взаимоотнош-я. Разработано большое кол-во программ для сравн-я послед-й с последующим опред-м их сходства, но наиболее часто исп-ся программы серии BLAST. Пакет программ BLAST. В этот пакет входят программы для нахожд-я локального выравн-я между заданной послед-ю и послед-ми из базы данных (БД). Его можно исп-ть как для случая ДНК, так и для белк-х последовательностей. Доступная версия программы находится на сервере NCBI. Сем-во программ серии BLAST можно разделить на 7 осн-х групп: 1. Геномные программы – предназн-ны для сравн-я изуч-й нуклеотидной послед-ти с БД секвенир-го генома (Arabidopsisthaliana, Oryzasativa и др.). 2. Нуклеотидные – предн-ны для сравн-я изуч-й нуклеот-й послед-ти с БД секвенир-х НК и их уч-в. Туда входят: • blastn – медленное сравн-е с целью поиска всех сходных послед-й; • megablast – быстрое сравн-е с целью поиска высоко сходных послед-й; • dmegablast – быстрый поиск очень похожих, но не идентичных послед-й; 3. Белковые – предн-ны для сравн-я изуч-й аминокис-й послед-ти белка с имеющейся БД белков и их уч-в. Туда входят алгоритмы: • blastp – медл-е сравн-е с целью поиска всех сходных послед-й; • psi–blast – сравн-е с целью поиска послед-й, облад-х незначит-м сходством; • phi–blast – поиск белков, сод-х опред-й пользователем паттерн и др. 4. blastx – сравн-т транслир-ю послед-ть с БД белковых послед-й.5. tblastn – сравн-т аминокисл-ю послед-ть с БД транслир-х нуклеот-х послед-й. 6. tblastx – сравн-т транслир-ю послед-ть ДНК с БД транслир-х нуклеот-х послед-й. 7. Специализированный: • cdart – сравн-е с целью поиска гомолог-х белков по доменной архитектуре; • VecScreen – опред-е сегм-в нуклеот-й послед-ти НК, которые могут иметь векторное происх-е и др.; • bl2seq – локальное выравн-е двух послед-й и др. Принцип работы BLAST. Сначала алгоритм BLAST создает таблицу всех «близких» слов фиксир-й длины (по умолчанию – длины 3 для белковых послед-й, 11 — для нуклеот-х), которые бы локально выравнивались с заданной послед-ю. При этом вес выравн-я должен быть выше некого порогового знач-я. Затем алгоритм сканирует БД, и всякий раз, когда находит слово из списка, начинает процесс «расширения совпадения», чтобы увел-ть возм-й участок выравн-я без разрывов, в обоих напр-х, до достижения максим-го веса. После этого вычисл-ся статистическая значимость найденных совпад-й, и если она превышает опред-й порог, то выдается рез-т. Рез-т поиска, например в blastn, включает в себя: 1) Графич-е изобр-е обнар-х гомологов; 2) Список гомологов с оценкой значимости находки; Основные веб-ресурсы по протеомике: Протеомика - наука, осн-м предм-м из-я кот-й явл-ся белки, их фу-и и взаимод-я в живых орг-х. Базы данных по протеомике. Сод-т инф-ю о протеоме или какого- нибудь организма (человек, мышь, дрозофила, дрожжи и т.д.), или о протеоме каких-то опред-х типов органелл, клеток, тканей (протеом ядра, протеом мышечных клеток, нервной ткани и т.д.). Напр: База данных «Протеомика рака простаты» сод-т около 300 белк-х фракций, База данных «Протеомика мышечных органов сод-т более 300 идентифицированных мышечных белков. 47. Закономерности эволюции белковых структур. Проблемные аспекты предсказания функций белка. Белки эволюцион-т благодаря изменениям, вызванным мутациями в генах, которые их кодируют, что приводит к изменениям аминокислотной послед-ти. Следствие мутаций - изменение в структуре. В ходе эволюции белки могут: сохранять функцию и специфич-ть;сохранять функцию, но менять специфич-ть;менять функцию на подобную или ту же, но в др-м метаболич-м «контексте»;переключ-ся на совершенно др функцию. В идеальном случае из послед-ти мы узнаем структуру, а затем из структуры — функцию белка. Но отнош-я между структурой и функцией сложные. Белки с одинаковой структурой и даже с одинаковой послед-ю могут использов-ся для разных функций. Очень сильно различ-ся белки могут выполнять одинаковые функции. Трудность аннотации белковых послед-й усугуб-ся отсут-м четкого определ-я понятия “функция белка”. Для молекул-го биолога функцией белка яв-ся уч-е в сети белок-белковых взаимодействий. Для биохим-ка функция опред-ся метаболич-м процессом, в котором уч-т белок. Физиолог под функцией белка может понимать тканевую специфич-ть и т.д.. Обычно понятие функции белка разделяют на две составляющ-е: молекуляр-ю (биохим-ю) функцию и контекстную функцию (роль белка в клетке). При этом под молекуляр-й функцией понимают связыв-е, активация, ингибир-е, катализ и т.д. Контекстная функция - система структурно-функц-х белок-белковых взаимодействий. Сущ-т 2 подхода для предсказ-я функции белков:1-й использует тот факт, что белки, имеющие сходные послед-ти аминокислот имеют схожую вторич-ю и 3-ую структуру и часто имеют сходную функцию в клетке. Если для белка с неизвестной функцией удается найти др-й белок, схожий на уровне послед-ти, то можно предположить что и первый белок выполняет ту же задачу. Такие методы предназначены для предсказ-я молекулярной функции белка.2-й - для предсказ-я функций белков использует данные сравнительной геномики, изучая встреч-ть генома и относител-е располож-е в др-х геномах. Этот метод позволяет группир-ть белки, несущие сходную функцион-ю нагрузку(ферменты, участв-е в одном метаболич-м пути, или субъединицы, входящие в состав белкового комплекса). 48. Протеом. Проблемы представления протеома на основе нуклеотидных сиквенсов. Сравнительная протеомика (примеры протеомов). Протеом — термин для обозначения всей совокупности белков орг-ма, производимых клеткой, тканью или организмом в определённый период времени. Или, более строго, это совокупность экспрессированных белков в данном типе клеток или в организме, в данный период времени при данных условиях. Термин является производным слова «протеин» (белок), аналогичным по происхождению слову «геном» (совокупность всех генов организма). Протеом, особенно у эукариот, больше, чем геном, то есть количество белков превышает количество генов. Это связано с альтернативным сплайсингом, а также с посттрансляционной модификацией белков, например, их гликозилированием и фосфорилированием. В то время как геном определяется последовательностью нуклеотидов, протеом не сводится к сумме последовательностей аминокислот. Протеом включает в себя также пространственные структуры всех содержащихся в нём белков (см. вторичная структура белка, третичная структура белков) и функционального взаимодействия между ними. Протеомика — наука о протеомах, развивалась в значительной степени путём разделения белков методом двумерного электрофореза. Первое измерение — разделение белков на основе их электрического заряда методом изоэлектрического фокусирования. Второе измерение — разделение белков на основе их молекулярного веса методом en:SDS-PAGE. Протеомика занимается каталогизацией и анализом белков с целью установления: 1)в каком сотоянии (и на каком этапе жизн.цикла) клетки экспрессируется данный белок; 2)в каком количестве синтезируется данный белок; 3)с какими другими белками может взаимодействовать данный белок. Протеом изменяется с течением времени и определяется как совокупность белков отдельного образца или экземпляра (ткань, организм, клеточная культура) в определённый момент времени. Протеомика, таким образом, отражает биологическую активность генома в динамике. Сравнительная протеомика. Сравнение протеомов двух организмов (необязательно близкородственных) позволяет выявить как общие для этих двух организмов белки, так и белки, которые обуславливают различия их фенотипов. Такой анализ может давать информацию, полезную для понимания эволюционного процесса, а иногда это позволяет определить раннее неизвестные функции белков. 49. Биоинформатика(БИ) и разработка лекарств. 1 из самых перспектив-х и быстро развивающ-ся областей БИ – констр-ние лек-в.Лек. препарат–это хим. вещ-во, мол-ла кот-го взаимод-т со специфич-й биологич-й молекул-й мишенью внутри орг-ма и из-за такого взаим-вия вызывает физиологич-й эффект. Молекул-е мишени отн-ся к белкам.Чтобы понять,как функц-т белок-мишень,надо знать его первич. аминокисл-ю последоват-ть и 3-хмерную стр-ру макромол-лы,установить место связыв-я лиганда.Рез-ты даёт сочет-е эксперимент-х методов молекул-й биологии с компьют.методами. Если пространств. стр-ра белка-мишени не известна, строят его 3-хмерную модель. Соврем. молекул. компьют. моделиров-е сост-т из 4-х осн-х этапов:1) компьют. графика – визуализ-ся пространств. располож-е атомов в мол-х.2) расчёты квантовой механики – позвол-т точно рассчит-ть стр-ру и электростатич. потенциалы для небол-х мол-л.3) расчёты молекул. механики – определ-е стр-ры белка.4) моделиров-е молекул. динамики–примен-ся для исследов-я внутримолекул-х движений и уточнения 3-хмерных стр-р белков.За опознаванием мишени идет процесс её утвержд-я.Процесс включ-т моделиров-е болезней на опытных жив-х и анализ данных об экспрессии генов и белков.Когда установлен ген, кот. в болезнен. состоянии обнаруж-т повышающ. или понижающ. регуляцию, определ-т его природу с помощ. методов БИ.Еще испол-т программу BLAST, проводят поиск подобных ему генов или белков в базах данных последоват-тей. Подобные гены и белки помогают опред-ть ф-цию подверж-го регуляции гена. Адекватная мишень должна обладать высоким терапевтич. показателем, т.е. д.б. гарантия существен-го терапевти ч-го эффекта при введ. такого препарата. Если мишенью явл-ся известный белок,то акт-ть к связыв-ю м.б. измерена непосред-но. Потенциал-й противомикр-й препарат м.б. испытан путём наблюд. его возд-я на рост кул-ры патог-х МО. После оконч-го утвержд-я мишени начин-ся поиск лидов, кот. взаимод-т с этой мишенью, а после обнаруж-я лида, он д.б. оптимизирован. После оптимизации лиды оценивают по кач-ву: лёгкость синтеза и приготовл-я лекарств-й формы. Затем оптим. соед-е регистр-т как новый разработ-й препарат и направл-т на клинич. испытания. Они призваны определить уровень безопас-и и переносимости препарата при леч. пациентов и опред-ть его метаболич. путь в орг-ме.БИ необходима не только при анализе последоват-тей и структур, но также и для построения алгоритмов моделир-я взаимодейс-й белка-мишени с молек-ми препарата. Так,стала возможной рационал. разр-ка препаратов, где на основании данных о стр-ре белка предсказ-т тип лигандов, взаимодействующ. с данной мишенью, и т.о. существенно сужают диапазон соед-й, среди кот-х провод-ся поиск лидов. 50. Базы данных метаболических путей и их связь с информацией о генах и белках. Банки данных метаболических путей. Банк данных KEGG (Kyoto Encyclopedia of Genes and Genomes) собирает отдель-ые геномы, продукты и функции генов, но его особое достоинство закл-ся в объединении биохим-ой и генет-ой инфор-и. KEGG сосредот-ся на взаимоде-ях: молекул-й сборке, метаболи-их и регулят-ых сетях. KEGG организует/систематизирует пять типов данных во всеобъемлю-щую/полную систему: 1. Каталоги хими-их структур живых клеток 2.Каталоги генов 3. Карты геномов 4. Карты путей 5. Таблицы ортологов. Каталоги хим-их соеди-й и генов (1 и 2) содержат информ-ю о конк-ых моле-ах и последов-ях. Геномные карты (3) объединяют инфор-ю о генах в соответствии с тем, как они следуют в хромосомах. В некот-х случаях знание о том, что ген входит в состав оперона, может дать ключ к пониманию его функции. Карты биохим-х путей (4) описывают сети взаимосвя-х молек-ых функций, как метабол-их, так и регуля-ых. Так, карта метаб-ого пути в KEGG — это идеализи-ое представление большого числа возможных реальных метабол-их каскадов различных организмов. Каждая карта может служить основой для воссоздания реального метаб-го пути конкретного организма. Для этого ферментам, обознач-им звенья в общей схеме, ставятся в соответствие конкретные белки данного организма. Конкретный фермент организма может быть найден в KEGG в таблице ортологов (5), которая устан-ает связь фермента с его родственниками в других организмах. Это позволяет анали-ать родство метабол-х путей из разных организмов. Основа эффективности KEGG закл-ся в очень плотной сети ссылок между этими типами инфор-и и в наличии дополн-ых ссылок на внешние источники, доступ к кот-м поддержи-ся системой. Два примера вопросов, ответы на которые можно получить с помощью KEGG.*Предпол-ся, что простые метабол-ие пути эволюцион-ют в бо-лее сложные благодаря удвоению генов и их послед-му расхождению. Поиск наборов сходных ферментов в каталоге биохим-х путей позво-ляет выявить кластеры паралогов.*KEGG позволяет проверить, укладывается ли опреде-ый набор ферментов какого-либо организма в одну из известных схем метаб-их путей. Пустое звено в схеме указывает на отсутствие в организме соответ-ствующего фермента либо на сущест-ие неизвестного альтернативного пути. |