1 Основные исторические вехи в развитии биоинформатики
 Скачать 1.58 Mb. Скачать 1.58 Mb.
|

|
База данных TGP (TransGene Promoters) является коллекцией экспериментально проверенных промоторов растений, использованных для экспрессии чужеродных генов в высших растениях. TGP содержит информацию о размере промотора, его нуклеотидной последовательности, паттерне транскрипции и регуляторах, влияющих на активность промотора. При описании промотора приводится название вида растений, использованных для трансгенеза, поскольку вид растения-хозяина влияет на тканеспецифичность и активность чужеродной трансгенной конструкции. Все промоторы в базе TGP сопровождаются ссылками на соответствующие публикации и базу последовательностей GenBank. Для промоторов указаны позиции относительно старта транскрипции или трансляции. EPD - Eukaryotic Promoter Database - база данных аннотированных эукариотических промоторов РНК-полимеразы II, для которых экспериментально определен сайт старта транскрипции. Информация привязана к промоторной последовательности путем указания номера позиции на последовательности в базе EMBL. Каждый документ содержит библиографические данные, ссылки на родственные базы данных и содержит информацию, касающуюся картирования инициирующих сайтов транскрипции. 18. Геномные проекты. Веб-ресурсы по полным геномам. Базы данных OMIM и OMIA. 1. GOLD – genome online database (содержит перечень орган-ов с полно-ю сиквенир-ми геномами, сведения о штамме, сорте и т.д., но не содежит полного сиквенса генома). По гиперссылкам можно выйти на банк данных NCBI, где содер-ся данные геномы в виде контигов. 2.NCBI – Genome - Search[Страница NCBI – Genomes and maps: a) перечень баз данных б) либо выбрать в ниспадающем меню NCBI search ‘Genome’ и набрать в строке поиска название организма.]. 3.UCSC Genome Browser website. Инстр-ы: 1)GenomeBrowser - Позволяет « прокручивать » карты хромосом онлайн , при разл-ом увел-и и с разл-и аннотациями . 2)TheGeneSorter Показывает гомологию между генами . Эта программа отображает в виде упорядо-ой таблицы генов , кот-е связаны друг с другом . Отношение может быть одним из неско-их типов , в том числе белков на уровне гомологии , сходства профилей экспрессии гена , или геномной близости 3)Blat-быстро отображает последов-сть в геноме . Помещает ваш сиквенс на генетич-ю карту. 4) Table Browser- обеспечивает удобный доступ к основной базе данных. 5) VisiGene 6) GenomeGraphs(ПЕРЕВОД С АНГЛ.!!).3.1. HumanGenome, ENCODE . Генет-ая карта функцио-ых элементов генома чел-а . ENCODE - это поисковая система, которая позволяет находить разл-ые элементы в геноме человека и мыши, а также файлы, в кот-х хра-ся сиквенсы. 3.2. Браузер мышиного генома на ENCODE - Mouse Genome 3.3. USCS браузер для микробных геномов (прокариоты) 4. Карты челове-их хромосом. NCBI – Human Genome Resources 5. Карты челов-х хромосом, на кото-х отмечено положение маркеров генетич-х заболе-ий - Human Genome Landmarks Poster: Chromosome Viewer 6. OMIM 7. Saccharomyces Genome. OMIA - On-line Mendelian Inheritance in Animals. База данных генов и фенов наследс-х болезней живо-х, для которых не существует обширных баз данных (т.е. всех, кроме человека, крысы и мыши). Эта база имеет начало в коллекции, кот-я назы-ся Mendelian Inheritance in Man (MIM) и создана доктором McKusick. OMIA создана в 1980 году и содержит ссылки на публикации, каса-ся всех особенностей и болезней, кот-е явл-ся семей-и и связаны с наследств-ю. Она не содержит последоват-ей. OMIM - On-line Mendelian Inheritance in Man. Обширная база данных челов-х генов и генет-х болезней, созданная доктором МакКасиком (Victor A. McKusick) с коллегами в центре медиц-ой генетики (Johns Hopkins University, Baltimore, USA), поддер-ся совместно с NCBI. Содержит текстовую инфор-ю, картинки, ссылки, а также перекрестные ссылки на базы данных ENTREZ. 19. Сиквенсы биомолекул и секвенирование: общие определения. Понятие о символах неопределенности для записи нуклеотидных последовательностей. Форматы данных для записи сиквенсов. Секвенирование биополимеров (Белков и НК) — определение их аминокислотной или нуклеотидн-й послед-ти. В резте секв-ния получают формальное описание первичной структуры линейной макромолекулы в виде послед-сти мономеров в текстовом виде. Размеры секвен-мых уч-в ДНК обычно не превышают 1000 пар нуклеотидов (по Сэнгеру). В рез-те секв-ния участков ДНК, пол-т послед-ти участков генов, целых генов, тотальной мРНК и даже полных геномов орг-мов . Нукл-ные послед-ти, которые прочитывают в различных лабораториях по всему миру, данные депозитируются в трех базах данных, доступ к которым открыт для всех через интернет.
2. EMBL Nucleotide Sequence Database 3. DDBJ: DNA Data Bank of Japan. Сиквенсы биомолекул, и сиквенсы ДНК м.б. написаны в различных текстовых форматах, в зав-ти от программы, которая их будет анализ-ть или от БД, в которой они будут храниться. Программы работы с сиквенсами обычно имеют конвертеры для преобразования форматов. Форматы записи сиквенсов делятся на 2 большие группы: слоистые (несколько записей продолжаются многократно, в каждой серии строк располагаясь в том же порядке, как в самой первой серии) и неслоистые (каждая отдельная запись идет друг за другом). Наиб. часто используемые форматы: 1) fasta (с ним мы работали на парах), выглядит так >xyz some other comment ttcctctttctcgactccatcttcgcggtagctgggaccgccgttcagtcgccaatatgc agctctttgtccgcgcccaggagctacacaccttcgaggtgaccggccaggaaacggtcg cccagatcaaggctcatgtagcctcactggagggcatt 2) GenBank format LOCUS HSFAU ACCESSION X65923 DEFINITION H.sapiens fau mRNA BASE COUNT 125 a 139 c 148 g 106 t 1 ttcctctttc tcgactccat cttcgcggta gctgggaccg ccgttcagtc gccaatatgc 61 agctctttgt ccgcgcccag gagctacaca ccttcgaggt gaccggccag gaaacggtcg 121 cccagatcaa ggctcatgta gcctcactgg agggcattgc cccggaagat caagtcgtgc 181 tcctggcagg cgcgcccctg gaggatgagg ccactctggg ccagtgcggg gtggaggccc 301 gtgctggaaa agtgagaggt cagactccta aggtggccaa acaggagaag agaagaaga 3) EMBL format ID HSFAU standard; DNA; UNC; 518 BP. AC X65923; DE H.sapiens fau mRNA SQ Sequence 518 BP; 125 A; 139 C; 148 G; 106 T; 0 other; ttcctctttc tcgactccat cttcgcggta gctgggaccg ccgttcagtc gccaatatgc 60 agctctttgt ccgcgcccag gagctacaca ccttcgaggt gaccggccag gaaacggt 120 cccagatcaa ggctcatgta gcctcactgg agggcattgc cccggaagat caagtcgt 180 tcctggcagg cgcgcccctg gaggatgagg ccactctggg ccagtgcggg gtggagg 240 tgactaccct ggaagtagca ggccgcatgc ttggaggtaa agttcatggt tccctggcc 300 4) Filip format 5) mega format 6) msf format 7) nexus / paup format. Для записи последовательностей ДНК и РНК, согласно правилам международной химической номенклатуры, приняты стандартные символы неопределенности для случаев, когда есть двусмысленность в чтении тех или иных нуклеотидов, полученная на секвенаторе. Четыре символа биологич-го алфавита были приняты еще в 1970 г. Все использующиеся символы неопределенности представлены в таблице: 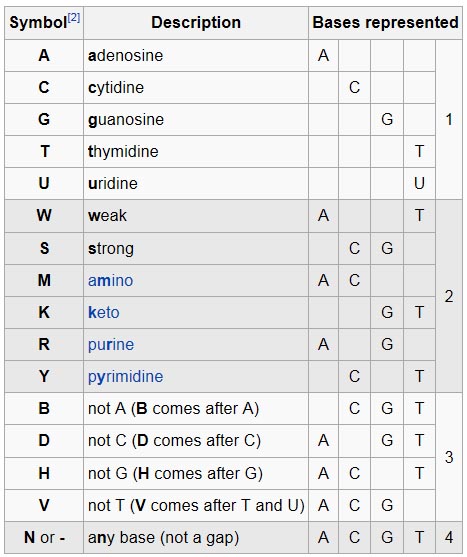 Напр H означает то, что данный нуклеотид не является гуанином, а R указывает на то, что в данном месте находится пуриновое основание (аденин или гуанин). 19.Секвенирование нуклеиновых кислот: история и классификация методов. Секвенирование ДНК – это процесс чтения последовательности нуклеотидных оснований в ее однонитевой молекуле. История. Несмотря на то, что большинство исторических очерков гласят, что впервые сообщение о методе сиквенирования ДНК появилось в 1977 г. (Максам, Гилберт и независимо Санждер), на самом деле первый ДНК-сиквенс был получен еще в начале 1970 г, крайне громоздким методом двумерной хроматографии (при которой подвижная фаза после прохождения через первую колонку, переносится во вторую колонку, но уже обладающую другими свойствами). В 1973 г. Гилберту и Максаму удалось сиквенировать фрагмент ДНК длиной 24 основания, крайне трудоемким методом блуждающих пятен. Как видим, уже на заре сиквенирования существовали различные лаораторные методы чтения ДНК. В период 1972–1976 бельгийский исследователь Walter Fiers и соавторы, работая с РНК, сиквенировал сначала отдельно взятый ген, а потом целый геном бактериофага MS2. Таким образом, чтение РНК-молекул и целых РНК-геномов шло несколько впереди чтения ДНК-сиквенсов. Ныне все методы сиквенирования делят на 3 группы: Первоначальные, или основные методы. * Сиквенирование по Максаму-Гилберту (химическое секвенирование) * Секвенирование по Санджеру (метод терминации цепи) II. Методы нового поколения. Методы нового поколения (next-generation) резко отличаются от первоначальных методов, во-первых, высокой производительностью, и во-вторых, более дешевы. * Массивное параллельное сиквенирование подписей - Massively Parallel Signature Sequencing (MPSS). * Полониевое секвенирование *454 пиросеквенирование * Метод секвенирования, предложенный фирмой Illumina (Solexa) *СОЛИД-секвенирование- SOLiD sequencing (2-основное раскодирование) * Ионно-полупроводниковое секвенирвоание * Секвенирование с помощью ДНК-наношариков * Секвенирование одной молекулы на секвенаторе Гелиоскоп * Секвенирование одной молекулы в реальном времени * Секвенирование с помощью РНК-полимеразы III. Методы, находящиеся в разработке. * Секвенирование при прохождении ДНК через нанопоры * Секвенирование путем гибридизации * Микрофлюидное секвенирование по Санджеру 21.Секвенирование ДНК по Сенгеру и его разновидности. Дезоксинуклеотидный метод, или метод «обрыва цепи», был разработан Ф. Сенгером в 1977 году и в наст. время широко испол-ся для опред-ния нуклеот-ой последоват-сти ДНК. При секвенировании по Сенгеру происх-т гибридизация синтети-го олигонуклеотида длиной 17-20 звеньев со специфи-им участком одной из цепей секвенир-го участка. Этот олигонуклеотид явл-ся праймером, поставляющим 3'-ОН группу для инициации синтеза цепи, комплемент-ой матрице.Раствор с праймером распред-ют по 4м пробиркам, в каж-й из кот-х нах-ся 4 дезоксинуклеотида,один из них-меченный радиоактивным изотопом и один из четырех 2',3'-дидезоксинуклеотидов. Дидезоксинуклеотид вкл-ся по всем позициям в смеси раст-х цепей, и после его присоед-ия рост цепи сразу останав-тся. В рез-те этого в каждой из 4х пробирок при участии ДНК-полимеразы образ-ся уникальный набор олигонукл-в разной длины, включ-х праймерную последов-сть. Далее в пробирки добавл-т формамид для расхождения цепей и проводят электрофорез в полиакрила-ом геле на 4х дорожках. Проводят радиоавтографию, к-я позвол-т «прочесть» нуклеот-ю последоват-сть секвенир-го сегмента ДНК. В более совреме-м варианте дидезоксинуклеотиды метят 4мя разными флуоресце-ми красителями и проводят ПЦР в одной пробирке. Затем во время электр-за в полиакриламидном геле луч лазера в определ-м месте геля возбуж-т флуоресценцию красителей, и детектор опред-т, какой нуклеотид в наст. момент мигрирует через гель. Соврем-ые приборы исп-т для секвенирования ДНК капиллярный электрофорез. 22. Подготовит-е этапы при высокопроизводительном секвенир-и: создание библиотеки и эмульсионная ПЦР. Пиросеквенирование ДНК. Первый этап – подготовка библиотеки одноцеп-х мол-л ДНК-матрицы. ДНК, сод-ся в пробе (например, геномная ДНК), фрагм-ся случ-м образом на короткие молекулы длиной 300 – 800 п.о. К каждой из цепей получ-х фрагментов лигируется два олигонуклеот-х адаптера: адаптер A и адаптер B. Второй этап – клональная амплиф-я каждого из фрагм-в ДНК методом эмульсионной ПЦР. Каждый из одноцеп-х фрагм-в иммобилиз-ся на полимерном шарике диаметром 28 мкм, несущем на пов-ти ковалентно связанные с ним праймеры, комплементарные послед-ти одного из адаптеров. Предвар-но смесь разъедененных на комплемент-е цепи фрагм-в разбавляют таким образом, было попадание на каждый из шариков только одной мол-лы ДНК. Каждый шарик с иммобилиз-м на нем фрагм-м ДНК заключ-ся в окруженную маслом микрокаплю ра-ра (водно-масляная эмульсия). Ра-р сод-т реактивы, необх-е для провед-я ПЦР, включая праймер, комплементарный послед-ти второго адаптера. Т.о, ПЦР-амлификация каждого фрагмента ДНК проводится внутри изолир-го микрореактора, что исключает контаминацию фрагм-ми ДНК с другой первичной структ-й. Третий этап – собств-но пиросеквенир-е. Эмульсия разруш-ся, синтезир-е в ходе ПЦР двухцеп-е молекулы ДНК разд-ся на комплемент-е цепи. Затем шарики, несущие одноцеп-е фрагм-ты ДНК помещ-ся в лунки спец-го планшета. Диаметр и высота лунки подобраны т.о., чтобы в каждую из них мог попасть только один шарик с ДНК-матрицей. Кроме того, в каждую лунку вносятся шарики меньшего диаметра с иммобилизованными на них ферментами, необходимыми для пирофосфатного секвенир-я – ATP-сульфурилазой и люциферазой светлячка. В лунки поступают реактивы необх-е для синтеза цепей, комплемент-х наход-ся на шариках. Каждый из четырех типов дезоксинуклеотидов, участв-х в реакции подается послед-но. При элонгации цепи на один нуклеотид происх-т выдел-е пирофосфата, который реагирует с аденозин 5'-фосфосульфатом (APS) и превращ-ся в ATP под действием сульфурилазы. АТР необходим люциферазе для окисления люциферина до оксилюциферина. В ходе окисл-я происх-т испускание кванта света (хемилюминесценция), кот-й детектируется отдельно для каждой лунки. Зарегистр-е сигналы дают инф-ю о послед-ти включения нуклеотидов в растущую цепь. Длина отд-го сиквенса составляет 240 – 400 п. о., всего за один раунд работы прибора анализир-ся до 100 миллионов п. о. Процесс пиросеквенир-я разработан Полом Нираном (Стокгольм, Швеция) в 1996 году. Основной принцип: в результате фермент-го превращения пирофосфата регистр-ся хемолюминисцентный сигнал, а высвобождение пирофосфата происх-т после встраивания необходимого нуклеотида. 23.Ионно-полупроводниковое секвенирование ДНК. Первичные диаграммы при разных видах секвенирования и процедура определения оснований (base calling). Ионно-полупроводниковое секвенирование - система, основанная на использ-и стандартной химии секвенир-я, но с системой обнаруж-я на полупроводниковой основе. Метод основан на обнаруж-и ионов водорода, которые выбрас-ся во время полимериз-и ДНК. Микропланшет, содержащей шаблонную нить ДНК, подлежащую секвенир-ю, залит одним типом нуклеотидов. Если введенный нуклеотид комплементарен к нуклеотидам шаблонной цепи, он включ-ся в растущую комплементар-ю цепь. Это приводит к высвобожд-ю иона водорода, который включает гипертензивный ионный датчик(указ-т, что реакция произошла). Если гомополимерные повторы присутствуют в матричной последов-ти, несколько нуклеотидов будут включены в единый цикл. Это приводит к соответ-му колич-ву выпущенных ионов водорода и пропорционально более выс-му электронному сигналу. Секвенир-е всегда содержит ошибки. Ошибки происходят на фазе, когда нужно распознать помеченные нуклеотиды (понять, каким цветом и с какой силой светятся кластеры из многократно клонир-х уча-в ДНК). Проблема - из-за неидеальности остальных этапов процесса кластеры никогда не светятся только одним цветом, это смесь 4-х цветов с той или иной интенсивностью. Нужно выделить наиболее интенсивную компоненту и оценить, насколько вероятна ошибка в этой букве. Эта задача наз-ся base calling (распознаван-е нуклеотидов). *Метод ионного полупроводн-го секвенир-я просто детектирует ионы, которые выдел-ся при присоедин-и нового нуклеотида к нити ДНК. Это позволяет сократить время и стоимость, хотя процент ошибок стан-ся больше. 24. Основные банки данных нуклеот-х последоват-тей. Структура записей для сиквенса нукл-й к-ты в генет-ом банке NCBI. Особенности в сиквенсе. Нукл-ные последов-ности, которые прочитывают в различных лабораториях по всему миру, депозитируются в 3 базах данных, доступ к которым открыт для всех через интернет. 1. GenBank. Архив возник на базе Национального центра биотехнол-ской информации (США). Это генетич-я база данных сиквенсов, аннотированная коллекция всех публично доступных сиквенсов ДНК. 2. EMBL Nucleotide Sequence Database. Этот архив нуклеотидных последоват-стей возник на базе Библиотеки данных (EMBL - European Molecular Biology Laboratory Data Library) Европейского института биоинформатики (Великобритания). Основные источники для последоват-стей ДНК и РНК являются прямыми материалами, представленными отдельными исследователями, проекты секвенирования генома и патентные заявки. База данных производится в рамках международного сотрудничества с GenBank (США) и базой данных ДНК Японии (DDBJ). Каждый из трех групп собирает часть общих данных о последовательносях, приведенных в мире, и все новые и обновленные записи базы данных, которыми обмениваются группы на ежедневной основе. 3. DDBJ: DNA Data Bank of Japan. Банк данных ДНК Японского национального института генетики. Единственный банк данных нуклеотидных последоват-стей в Азии, который официально сертифицирован для сбора нуклеот-х последоват-ей от исследователей, и имеющий международно признанный инвентарный номер в представлении данных. 1+2+3. The virtually unified database is called "INSD; International Nucleotide Sequence Database" Формат хранения данных и характер аннотаций сиквенсов (т.е. формат записей) между банками данных 1, 2 и 3 несколько различается. Но, благодаря ежедневному обмену данными, эти три крупнейших архива хранят сходный набор архивных записей расшифрованных нуклеотидных последовательностей. В документе присутствует описательная часть. Она помогает пользователям найти интересующую его последовательность и хранит дополнительную информацию, например, кто проводил эксперимент по сиквенированию, откуда взят образец, биологическая роль данной последовательности и т.д. Таблицы особенностей (features) – это составная часть аннотации сиквенса, которые описывают свойства специфических участков или отдельных кодирующих последовательностей внутри сиквенса.  |