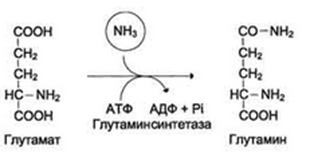
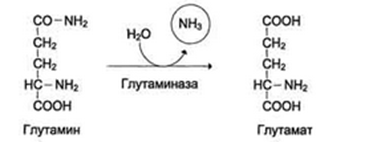
вопрос Роль белков в питании. 1 вопрос Роль белков в питании, нормы, азотистый баланс, коэффициент изнашивания, физиологический белковый минимум. Белковая недостаточность
 Скачать 1 Mb. Скачать 1 Mb.
|

Часть аммиака образуется в кишечникев результате действия бактерий на пищевые белки (гниение белков вкишечнике) и поступает в кровь воротной вены. Концентрация аммиака в крови воротной вены существенно больше, чем в общем кровотоке. В печени задерживается большое количество аммиака, что поддерживает низкое содержание его в крови. Концентрация аммиака в крови в норме редко превышает 0,4-0,7 мг/л (или 25-40 мкмоль/л90% выводятся почками.Аммиак легко проникает через мембраны в клетки и в митохондриях сдвигает реакцию, катализируемую глутаматдегидрогеназой, в сторону образования глугамата:  Рис. 9-10. Метаболизм азота глутамина в кишечнике. В почках также происходит гидролиз глутамина под действием глутаминазы с образованием аммиака. Этот процесс является одним из механизмов регуляции кислотно щелочного равновесия в организме и сохранения важнейших катионов для поддержания осмотического давления. Глутаминаза почек значительно индуцируется при ацидозе, образующийся аммиак нейтрализует кислые продукты обмена и в виде аммонийных солей экскретируется с мочой (рис. 9-11). Эта реакция защищает организм от излишней потери ионов Na+ и К+, которые также могут использоваться для выведения анионов и утрачиваться. При алкалозе количество глутаминазы в почках снижается. В почках образуется и выводится около 0,5 г солей аммония в сутки. Высокий уровень глутамина в крови и лёгкость его поступления в клетки обусловливают использование глутамина во многих анаболических процессах. Глутамин - основной донор азота в организме. Амидный азот глутамина используется для синтеза пуриновых и пиримидиновых нуклеотидов, аспарагина, аминосахаров и других соединений (рис. 9-12). Рис. 9-11. Метаболизм амидного азота глутамина в почках.  Рис. 9-12. Пути использования глутамина в организме. В мозге и некоторых других органах может протекать восстановительное аминирование α-кетоглутарата под действием глутаматдегидрогеназы, катализирующей обратимую реакцию. Однако этот путь обезвреживания аммиака в тканях используется слабо, так как глутаматдегидрогеназа катализирует преимущественно реакцию дезаминирования глутамата. Хотя, если учитывать последующее образование глутамина, реакция выгодна для клеток, так как способствует связыванию сразу 2 молекул NH3. 15. Синтез мочевины. Орнитиновый цикл. Субклеточная локализация. Энергетика процесса. Гипераммониемии. Виды. Причины. Симптомы протекания заболевания. Метод количественного определения мочевины в сыворотке крови.  16 Метаболизм фенилаланина и тирозина. Нарушение обмена фенилаланина и тирозина: альбинизм, алкаптонурия, фенилкетонурия. Реакция качественного обнаружения фенилпирувата в моче. Метаболические дефекты при классической и атипичной фенилкетонурии. Основные проявления, терапевтическая тактика. Нарушения обмена этих АК связано с нарушением биосинтеза некоторых ферментов, которые катализируют метаболические превращения этих АК. Результатом нарушения синтеза ферментов является возникновение наследственных генетических заболеваний: 1) фенилкетонурия - нарушен синтез фенилаланин-гидроксилазы, поэтому фенилаланин превращается в фенилпируват, который оказывает токсическое воздействие на развитие некоторых отделов головного мозга. 2) альбинизм - нарушен синтез ферментов, превращающих ДОФА в ДОФА-хром, поэтому нарушается синтез меланинов. 3) алкаптонурия - нарушен синтез диоксигеназы гомогентизиновой кислоты, она выделяется с мочой, моча приобретает черный цвет. 4) кретинизм - нарушен синтез йодиназы, что приводит к нарушению синтеза йодсодержащих гормонов щитовидной железы. 5) может быть нарушен синтез фермента тирозиназы, который катализирует превращение тирозина в ДОФА, следовательно будет нарушаться синтез гормонов мозгового слоя надпочечников и меланина. Из всех этих заболеваний в настоящее время удается лечить фенилкетонурию, для этого из рациона ребенка исключают фенилаланин и увеличивают в пище количество тирозина. Если ребенка держать на этой диете до 6-7 лет, тогда не возникает умственная отсталость, т.к. к 6-7 годам успевают развиться отделы головного мозга, развитие которых задерживается при избытке в ткани мозга фенилпирувата.  16 Метаболизм фенилаланина и тирозина. Нарушение обмена фенилаланина и тирозина: альбинизм, алкаптонурия, фенилкетонурия. Реакция качественного обнаружения фенилпирувата в моче. Метаболические дефекты при классической и атипичной фенилкетонурии. Основные проявления, терапевтическая тактика. Нарушения обмена этих АК связано с нарушением биосинтеза некоторых ферментов, которые катализируют метаболические превращения этих АК. Результатом нарушения синтеза ферментов является возникновение наследственных генетических заболеваний: 1) фенилкетонурия - нарушен синтез фенилаланин-гидроксилазы, поэтому фенилаланин превращается в фенилпируват, который оказывает токсическое воздействие на развитие некоторых отделов головного мозга. 2) альбинизм - нарушен синтез ферментов, превращающих ДОФА в ДОФА-хром, поэтому нарушается синтез меланинов. 3) алкаптонурия - нарушен синтез диоксигеназы гомогентизиновой кислоты, она выделяется с мочой, моча приобретает черный цвет. 4) кретинизм - нарушен синтез йодиназы, что приводит к нарушению синтеза йодсодержащих гормонов щитовидной железы. 5) может быть нарушен синтез фермента тирозиназы, который катализирует превращение тирозина в ДОФА, следовательно будет нарушаться синтез гормонов мозгового слоя надпочечников и меланина. Из всех этих заболеваний в настоящее время удается лечить фенилкетонурию, для этого из рациона ребенка исключают фенилаланин и увеличивают в пище количество тирозина. Если ребенка держать на этой диете до 6-7 лет, тогда не возникает умственная отсталость, т.к. к 6-7 годам успевают развиться отделы головного мозга, развитие которых задерживается при избытке в ткани мозга фенилпирувата. 17 Общая схема синтеза гема. Нарушения синтеза гема-порфирии. Интоксикация свинцом. 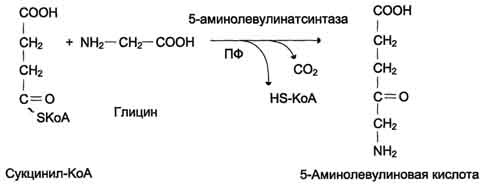 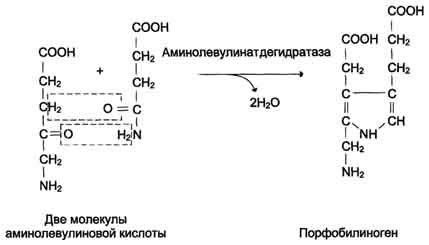 Гидроксиметилбилан-уропорфириноген III-копропорфириноген III-протофибрин IХ-гем Нарушения биосинтеза гема. Порфирии Наследственные и приобретённые нарушения синтеза гема, сопровождающиеся повышением содержания порфириногенов, а также продуктов их окисления в тканях и крови и появлением их в моче, называют порфириями . При этих заболеваниях отмечают снижение образования гема. Поскольку гем - аллостерический ингибитор аминолевулинатсинтазы, то активность этого фермента повышается, и это приводит к накоплению промежуточных продуктов синтеза гема - аминолевулиновой кислоты и порфириногенов. В зависимости от основной локализации патологического процесса различают печёночные и эритропоэтические наследственные порфирии. Эритропоэтические порфирии сопровождаются накоплением порфиринов в нормобластах и эритроцитах, а печёночные - в гепатоцитах. При тяжёлых формах порфирии наблюдают нейропсихические расстройства, нарушения функций РЭС, повреждения кожи. Порфириногены не окрашены и не флуоресцируют, но на свету они легко превращаются в порфирины. Последние проявляют интенсивную красную флуоресценцию в ультрафиолетовых лучах. 18. Регуляция синтеза гема. Регуляция активности и скорости синтеза фермента АЛК-синтазы количеством гема, железа и лекарственными препаратами. Регуляторную реакцию синтеза гема катализирует пиридоксальзависимый фермент аминолевулинатсинтаза. Скорость реакции регулируется аллостерически и на уровне трансляции ферментаАллостерическим ингибитором и корепрессором синтеза аминолевулинатсинтазы является гем В ретикулоцитах синтез этого фермента на этапе трансляции регулирует железо. На участке инициации мРНК, кодирующей фермент, имеется  Рис. 13-5. Регуляция синтеза гема и гемоглобина. Гем по принципу отрицательной обратной связи ингибирует аминолевулинатсинтазу и аминолевулинатдегидратазу и является индуктором трансляции α- и β-цепей гемоглобина. последовательность нуклеотидов, которая называется железочувствительным элементом. При высоких концентрациях железа в клетках оно образует комплекс с остатками цистеина регуляторного железосвязывающего белка. При низких концентрациях железа трансляция аминолевулинатсинтазы тормозится . Аминолевулинатдегидратаза также аллостерически ингибируется гемом, Дефицит пиридоксальфосфата и лекарственные препараты, которые являются его структурными аналогами, снижают активность аминолевулинатсинтазы. 19 Общая схема распада гема. «Прямой» и «непрямой» билирубин, клиническое значение его определения. Гем(гемоксигеназа)-биливердин(биливердинредуктаза)-билирубин(УДФ-глюкуранилтрансфераза)-билирубинмоноглюкуронид(УД-глюкуронилтрансфераза)-билирубиндиглюкуронид 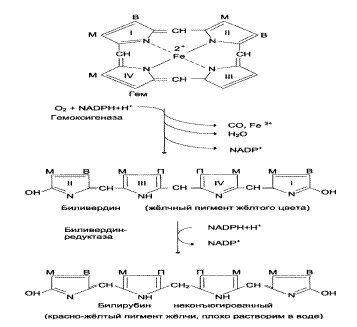 В нормальном состоянии концентрация общего билирубина в плазме составляет 0,3-1 мг/дл (1,7-17 мкмоль/л), 75% от общего количества билирубина находится в неконъюгированной форме (непрямой билирубин). В клинике конъ-югированный билирубин называют прямым, потому что он водорастворим и может быстро взаимодействовать с диазореагентом, образуя соединение розового цвета, - это прямая реакция Ван дер Берга. Неконъюгированный билирубин гидрофобен, поэтому в плазме крови содержится в комплексе с альбумином и не реагирует с диазореактивом до тех пор, пока не добавлен органический растворитель, например этанол, который осаждает альбумин. Неконъюгированный илирубин, взаимодействующий с азокрасителем только после осаждения белка, называют непрямым билирубином. Когда содержание билирубина превышает норму, говорят о гипербилирубинемии. В зависимости от того, концентрация какого типа билирубина повышена в плазме - неконъюгированного или конъюгированного, - гипербилирубинемию классифицируют как неконъюгированную и конъюгированную. У больных с печёночно-клеточной патологией, сопровождающейся длительным повышением концентрации конъюгированного билирубина, в крови обнаруживают третью форму плазменного билирубина, при котором билирубин ковалентно связан с альбумином, и поэтому его невозможно отделить обычным способом. В некоторых случаях до 90% общего содержания билирубина крови может находиться в этой форме. 20. Желтухи. Виды, причины возникновения. Количественное определение содержания «общего» и «прямого» билирубина в сыворотке крови (норма, принцип метода, расчет). Причинами гипербилирубинемии могут быть увеличение образования билирубина, превышающее способность печени экскретировать его, или повреждение печени, приводящее к нарушению секреции билирубина в жёлчь в нормальных количествах. Гипербилирубинемию отмечают также при закупорке желчевыводящих протоков печени. Во всех случаях содержание билирубина в крови повышается. При достижении определённой концентрации он диффундирует в ткани, окрашивая их в жёлтый цвет. Пожелтение тканей из-за отложения в них билирубина называют желтухой. Клинически желтуха может не проявляться до тех пор, пока концентрация билирубина в плазме крови не станет выше 50 мкмоль/л. 1. Гемолитическая (надпечёночная) желтуха результат интенсивного гемолиза эритроцитов. Она обусловлена чрезмерным образованием билирубина, превышающим способность печени к его выведению. Гемолитическая желтуха развивается при исчерпании резервных возможностей печени. Основная причина надпечёночной желтухи - наследственные или приобретённые гемолитические анемии. Гипербилирубинемия у больных гемолитической желтухой обусловлена значительным повышением (103 - 171 мкмоль/л) в крови концентрации непрямой билирубин). Образование в печени и поступление в кишечник больших количеств билирубинглюкуронидов (прямой билирубин) ведёт к усиленному образованию и выделению с калом и мочой уробилиногенов и более интенсивной их окраски Один из главных признаков гемолитической желтухи - повышение содержания в крови (непрямого) билирубина. Это позволяет легко отличить её от механической (подпечёночной) и печёночно-клеточной (печёночной) желтух. Неконъюгированный билирубин токсичен. Гидрофобный, липофильный неконъюгирован-ный билирубин, легко растворяясь в липидах мембраны и проникая вследствие этого в митохондрии, разобщает в них дыхание и окислительное фосфорилирование, нарушает синтез белка, поток ионов калия через мембрану клетки и органелл. Это отрицательно сказывается а состоянии ЦНС, вызывая у больных ряд характерных неврологических симптомов. |