ОБМЕН БЕЛКОВ
Фонд свободных аминокислот организма составляет около 30 г. Содержание аминокислот в крови равно 25-65 мг/дл. Источником свободных аминокислот организма служат пищевые белки, белки собственных тканей, а также синтез аминокислот из углеводов.
Значение аминокислот для организма определяется, прежде всего, тем, что они используются для синтеза белков. Кроме того, из аминокислот образуется большое количество азотсодержащих веществ небелковой природы, выполняющих специальные функции: биогенные амины, глутатион, гем и др. Катаболизм аминокислот может служить источником энергии для синтеза АТФ. При обычном питании энергетическая роль аминокислот невелика (10%), однако может быть существенной при преимущественно белковом питании, а также при голодании.
Азотистый баланс
На долю аминокислот (свободных и в составе белков) приходится более 95% всего азота организма. Поэтому об общем состоянии аминокислотного и белкового обмена можно судить по азотистому балансу, т.е. разнице между количеством азота, поступающего с пищей, и количеством выделяемого азота (главным образом в составе мочевины). У взрослого здорового человека при нормальном питании имеет место азотистое равновесие, т.е. количество выделяемого азота равно количеству поступающего. В период роста организма, а также при выздоровлении после тяжелых заболеваний азота выводится меньше, чем поступает, т.е. имеет место положительный азотистый баланс. При старении, голодании и в течение истощающих заболеваний азота выводится больше, чем поступает, т.е. имеет место отрицательный азотистый баланс.
При положительном азотистом балансе часть аминокислот пищи задерживается в организме, включаясь в состав белков и клеточных структур. Общая масса белков в организме при этом увеличивается. Наоборот, при отрицательном азотистом балансе общая масса белков уменьшается (катаболические состояния).
Переваривание белков в желудочно-кишечном тракте
Переваривание белков начинается в желудке под действием пепсина. Пепсин в виде неактивного предшественника - пепсиногена - образуется в главных клетках желудочных желез. Активация пепсиногена происходит в желудочном соке и заключается в отщеплении N-концевого фрагмента молекулы. В результате ограниченного протеолиза и последующей конформационной перестройки оставшейся части молекулы формируется активный центр, т.е. образуется пепсин. Превращение пепсиногена в пепсин может происходить под действием HCI (этот процесс протекает медленно) или самого пепсина, т.е. аутокаталитически (этот процесс протекает быстро). Т.о., небольшое количество пепсина, образовавшегося при участии HCI вскоре после секреции желудочного сока, быстро приводит к превращению остальной части пепсиногена в пепсин.
Поскольку пепсин относится к эндопептидазам, т.е. гидролизует пептидные связи внутри белковой молекулы, в результате его действия белки в желудке распадаются на полипептиды. Наибольшую активность пепсин проявляет при pH 1,5-2,5.
НС1, помимо активации пепсиногена, выполняет и другие важные функции. В кислой среде желудочного сока большинство белков денатурирует, что облегчает их переваривание. Кроме того, кислый желудочный сок обладает бактерицидным действием, препятствуя развитию микрофлоры в желудке.
Из желудка крупные осколки белков поступают в тонкий кишечник, в верхних отделах которого завершается переваривание белков. Это происходит под действием ферментов поджелудочной железы и клеток кишечника. В клетках поджелудочной железы синтезируются проферменты трипсиноген, химотрипсиноген,
прокарбоксипептидазы А и В, проэластаза. Активация трипсиногена происходит при участии фермента энтеропептидазы, выделяемого клетками кишечника. В результате отщепления N-концевого гексапептида и изменения конформации оставшейся части молекулы формируется активный центр и образуется трипсин. Все другие проферменты поджелудочной железы активируются трипсином путем ограниченного протеолиза. В результате получаются ферменты химотрипсин, карбоксипептидазы А и В, эластаза.
Трипсин, химотрипсин, эластаза подобно пепсину относятся к эндопептидазам и различаются по субстратной специфичности. Основную часть продуктов действия этих ферментов составляют пептиды.
Карбоксипептидазы - это экзопептидазы: они гидролизуют пептидную связь, образованную С-концевым аминокислотным остатком. Карбоксипептидаза А отщепляет преимущественно С-концевые аминокислоты с гидрофобным радикалом, а карбоксипептидаза В - С-концевые остатки лизина и аргинина.
Последний этап переваривания происходит при участии ферментов, синтезируемых клетками кишечника - аминопептидаз и дипептидаз. Аминопептидазы отщепляют N-концевые аминокислоты от пептидов, а дипептидазы гидролизуют дипептиды.
Последовательность действия всего набора протеолитических ферментов обеспечивает расщепление белков до аминокислот. Образовавшиеся аминокислоты всасываются слизистой кишечника при участии целого ряда механизмов, основным из которых является активный вторичный транспорт.
Обмен белков в тканях
Белки организма человека постоянно обновляются. В норме у здорового взрослого человека обновление белков составляет 1-2% от общего количества белков тела за сутки и связано преимущественно с деградацией мышечных белков до аминокислот. При этом примерно 75-80% высвободившихся аминокислот повторно используется в синтезе белков. Оставшаяся часть метаболизируется до конечных продуктов азотистого обмена, удаляемых из организма, а также превращается в глюкозу, кетоновые тела и СО2. Суточная деградация белков составляет 30-40 г. Поскольку
16% массы белка приходится на азот, суточная потеря азота составляет 5-7 г.
Распад белка в тканях происходит под действием тканевых протеиназ или катепсинов, локализованных преимущественно в лизосомах (pH 5-6). В зависимости от структуры активного центра ферментов, все катепсины подразделяются на
тиоловые (катепсины В, С, Н, L, N, S)
аспартильные (катепсин D)
сериновые (катепсин А)
Под действием катепсинов тканевые белки расщепляются до отдельных аминокислот.
Катаболизм аминокислот
Аминокислоты, поступающие в организм в количествах, превышающих потребности, связанные с биосинтезом белков, запасаться в организме не могут и используются как метаболическое топливо.
Катаболизм аминокислот чаще всего начинается с реакции дезаминирования - удаления а-аминогруппы. Дезаминированию подвергаются все аминокислоты, кроме лизина.
Различают следующие виды дезаминирования:
а) окислительное - для Glu
б) неокислительное - для Ser, Thr, His, Cys.
в) непрямое - для всех остальных аминокислот.
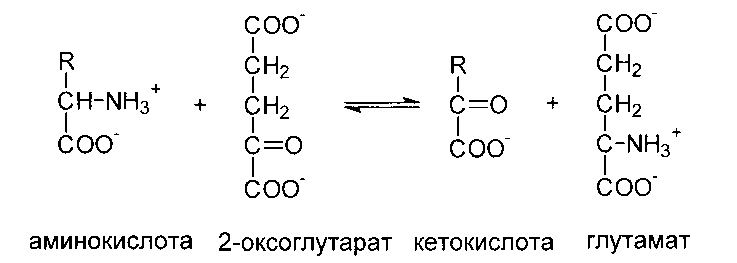
Непрямое дезаминирование аминокислот протекает в 2 стадии. Первая стадия - реакция переаминирования. Реакциями переаминирования называют реакции переноса аминогруппы от аминокислоты на кетокислоту с образованием новой аминокислоты и новой кетокислоты, а ферменты, катализирующие эти реакции, получили название аминотрансфераз или трансаминаз. Основным акцептором (сборщиком) аминогрупп является 2-оксоглутарат.
Кроме того, в роли промежуточных акцепторов аминогрупп могут выступать пируват и оксалоацетат. Образующиеся при этом, соответственно, аланин и аспартат могут передавать свою аминогруппу на 2-оксоглутарат с образованием глутамата. Таким образом, аминогруппы большинства аминокислот собираются в составе глутамата.
Вторая стадия: собственно дезаминирование. Реакция катализируется глутаматдегидрогеназой (ГДГ) - митохондриальным ферментом, использующим в качестве кофермента НАД+. Реакция обратима и функционирует как в процессах катаболизма, так и биосинтеза аминокислот.
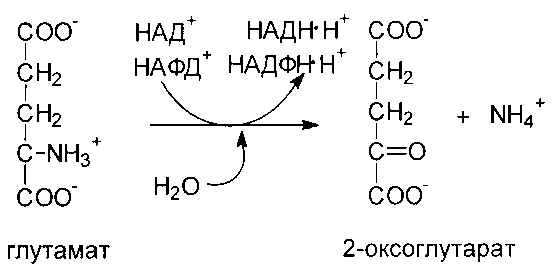
ГДГ печени является регуляторным ферментом. ГТФ и АТФ являются аллостерическими ингибиторами, тогда как ГДФ и АДФ служат аллостерическими активаторами. Следовательно, снижение энергетического заряда ускоряет окисление аминокислот.
Определение активности АлАТ в сыворотке крови широко используется в клинике с целью ранней диагностики и мониторинга заболеваний печени.
Обмен аммиака: источники, связывание в тканях, транспорт
Другими (помимо аминокислот) источниками аммиака в организме человека являются:
глутамин + Н2О → глутамат + NH3
амины (R-CH2-NH2 + Н2О + О2 —> R-С=О + NH3 + Н2О2)
аденин + Н2О → гипоксантин + NH3
распад пиримидиновых оснований:
3-уреидопропионат + Н2О → β-аланин + СО2 + NH3
Образовавшийся аммиак - вещество крайне токсичное, особенно опасное для мозга. Причины токсичности аммиака:
а) аммиак сдвигает реакцию, катализируемую глутаматдегидрогеназой в сторону образования глутамата
2-оксоглутарат + НАДН·Н+ + NH3 → глутамат + НАД+
Снижение концентрации 2-оксоглутарата вызывает угнетение обмена аминокислот (переаминирования) и гипоэнергетическое состояние (угнетение ЦЛК).
б) аммиак усиливает синтез глутамина из глутамата в нервной ткани
глутамат + NH3+ АТФ → глутамин + АДФ + Н3РО4
Накопление глутамина в нервных клетках приводит к повышению осмотического давления и в больших концентрациях может вызвать отек мозга. Снижение концентрации глутамата нарушает обмен нейромедиаторов, в частности синтез γ-аминомасляной кислоты (ГАМК) - основного тормозного медиатора.
Глутамат → ГАМК + СО2
Это приводит к преобладанию процессов возбуждения над процессами торможения и вызывает судороги.
в) Аммиак в крови и цитозоле образует ион NH4+
Глутаминсинтетаза обладает высоким сродством к аммиаку, и благодаря этой реакции в крови и тканях поддерживается низкая концентрация NH3.
Глутамин является транспортной формой аммиака, т.к. представляет собой нейтральную кислоту, способную легко проникать через клеточные мембраны путем облегченной диффузии (в отличие от глутаминовой кислоты, требующей механизмов активного транспорта).
Образовавшийся в тканях глутамин транспортируется в почки и печень.
Образование и экскреция аммиака в почках
Экскреция аммиака с мочой в норме невелика - около 0,5 г в сутки. Но при ацидозе она повышается в несколько раз. Аммиак в почках в основном образуется из глутамина при гидролизе последнего глутаминазой, имеющейся в клетках эпителия канальцев почек. Ацидоз стимулирует синтез глутаминазы, а также поглощение глутамина клетками почек; образующийся аммиак нейтрализует кислоты:
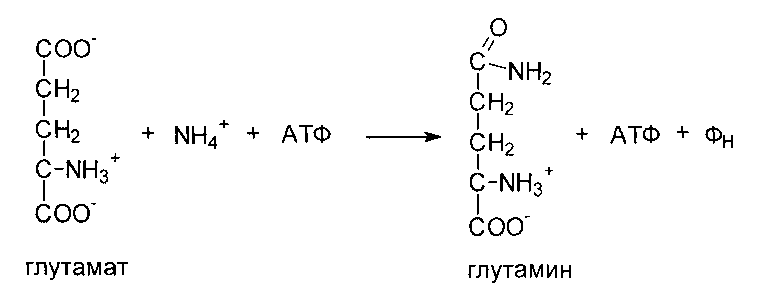
Накопление NH4+ нарушает трансмембранный перенос ионов, в частности одновалентных катионов Na+ и К+, что также влияет на проведение нервных импульсов.
Образовавшийся в клетках аммиак связывается (обезвреживается) и выводится из организма почками в виде конечных продуктов азотистого обмена: 1) мочевины - синтезируется в печени и 2) аммонийных солей - образуются в почках.
Существует несколько способов обезвреживания и выведения аммиака в разных тканях. 1) Образование глутамата из 2-оксоглутарата катализируется глутаматдегидрогеназой (ГДГ). Вклад этой реакции в обезвреживание аммиака невелик. 2) Основной реакцией обезвреживания аммиака почти во всех тканях является синтез глутамина под действием глутаминсинтетазы:
Образовавшаяся аммонийная соль выводится из организма. Таким образом, экскреция аммиака почками при ацидозе служит для выведения именно кислот, а не азота. Этот процесс обеспечивает сбережение организмом ионов Na+, которые в отсутствие ионов аммония выводились бы с анионами кислот.
Биосинтез мочевины
Мочевина - основной конечный продукт азотистого обмена, в составе которого из организма выводится избыток аммиака.
Катаболизм аминокислот и образование аммиака происходит во многих тканях. Для транспорта аммиака из тканей в печень используются 3 соединения: глутамин, аланин и аммиак.
Биосинтез мочевины происходит только в печени, т.к. только в этом органе имеется полный набор ферментов орнитинового цикла.
Один из двух атомов азота мочевины включается в нее за счет использования аммиака. При действии карбамоилфосфатсинтетазы I образуется карбамоилфосфат.
Цитруллин реагирует с аспарагиновой кислотой, превращаясь в аргининоянтарную кислоту, которая распадается на аргинин и фумаровую кислоту.
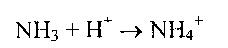
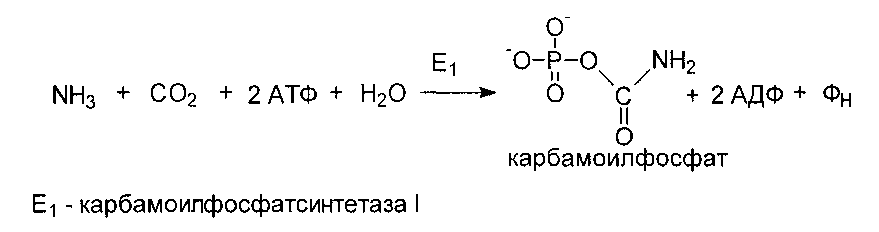
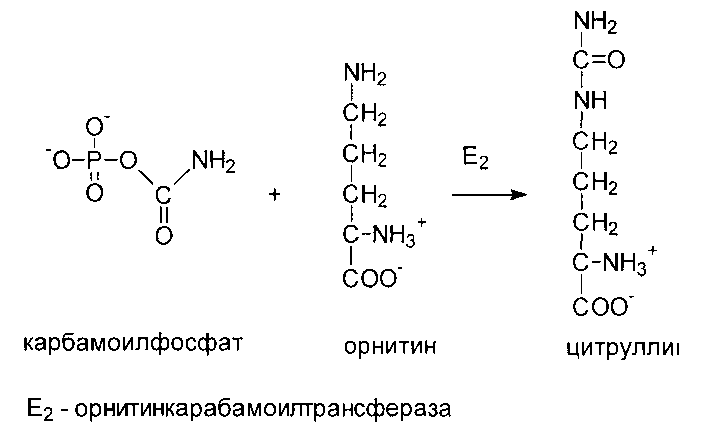
Карбамоильная группа далее переносится на орнитин с образованием цитруллина.
Реакции орнитинового цикла до стадии образования цитруллина проходят в митохондриях, а последующие стадии - в цитозоле. Суммарное уравнение синтеза мочевины:

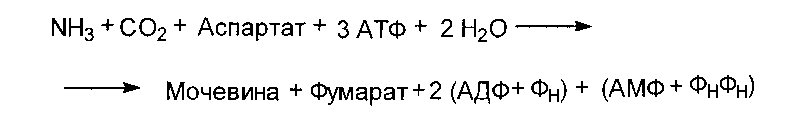
Биологическая роль синтеза мочевины
Орнитиновый цикл в печени выполняет 2 функции: 1) превращение азота аминокислот в мочевину, которая экскретируется и предотвращает накопление токсичных продуктов, главным образом аммиака; 2) синтез аргинина и пополнение его фонда в организме.
Регуляция биосинтеза мочевины
Ключевым ферментом биосинтеза мочевины является карбамоилфосфатсинтетаза
локализованная в митохондриях. Этот фермент - регуляторный; положительным модулятором для него служит N-ацетилглутамат.
Энергетическая цена синтеза мочевины
На синтез одной молекулы мочевины расходуется 4 высокоэнергетические фосфатные связи. 2 молекулы АТФ требуются для образования карбамоилфосфата и одна - для образования аргининосукцината. Однако в последней реакции АТФ претерпевает пирофосфатное расщепление, продуктами которого являются АМФ и пирофосфат, гидролизующийся затем с образованием двух молекул ортофосфата. Поэтому в общей сложности на образование одной молекулы мочевины расходуется 4 молекулы АТФ.
Г ипераммониемия
Диагностированы наследственные нарушения, вызываемые блокированием одной из реакций цикла мочевины. Общим признаком этих нарушений является повышенное содержание NH4+ в крови - гипераммониемия. Почти полная недостаточность какого-либо фермента цикла мочевины вызывает кому и приводит к смерти вскоре после рождения. Частичная недостаточность этих ферментов вызывает задержку умственного развития, временами рвоту. При мягких формах этих наследственных нарушений малобелковая диета и введение метаболитов орнитинового цикла (аргинина, цитруллина, глутамата) приводит к снижению содержания аммиака в крови и к улучшению клинической картины.
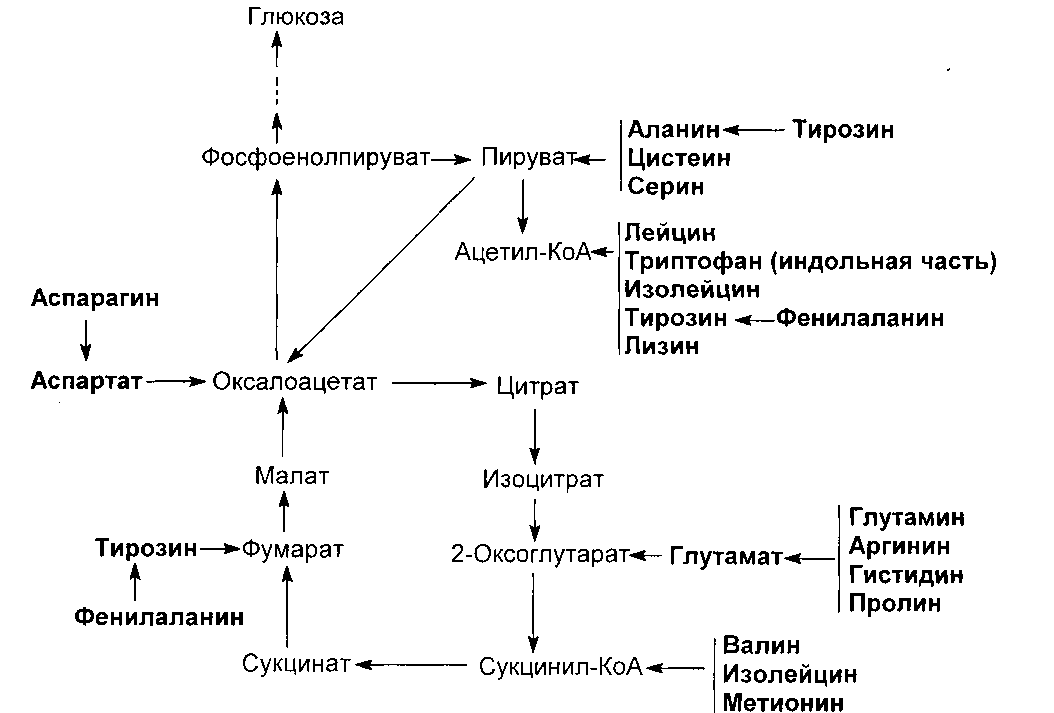
Судьба углеродных скелетов аминокислот
Стратегия разрушения аминокислот состоит в направленном превращении в
главные промежуточные продукты обмена веществ (пируват, ацетил-КоА, ацетоацетил-КоА, 2-оксоглутарат, сукцинил-КоА, фумарат и оксалоацетат), которые далее превращаются в глюкозу или окисляются в ЦЛК. Аминокислоты, распадающиеся с образованием ацетил-КоА или ацетоацетата, называются кетогенными, поскольку в результате распада повышается содержание кетоновых тел. К ним относится лейцин. Аминокислоты, распад которых приводит к образованию глюкозы, называются гликогенными. Возможность синтеза глюкозы из этих аминокислот обеспечивается тем обстоятельством, что указанные метаболиты ЦЛК и пируват могут превращаться в фосфоенолпируват и затем - в глюкозу. Образование глюкозы из аминокислот стимулирует гормон кортизол, который стимулирует синтез ферментов глюконеогенеза в печени. Ряд аминокислот используется и для синтеза глюкозы, и для синтеза кетоновых тел, так как в процессе катаболизма расщепляются с образованием двух продуктов, один из которых является метаболитом ОПК, а другой - ацетил-КоА или ацетоацетатом (лизин, изолейцин, фенилаланин, тирозин, триптофан). Такие аминокислоты называют смешанными или гликокетогенными.
Биосинтез заменимых аминокислот
Углеродный скелет 9 заменимых аминокислот (аланина, аспартата, аспарагина, глицина, серина, глутамата, глутамина, пролина, цистеина) может синтезироваться из глюкозы.
а-Аминогруппа вводится в соответствующие аминокислоты с помощью реакции переаминирования. Универсальным донором а-аминогруппы является глутамат.
Аспарагин синтезируется из аспартата и глутамина под действием аспарагинсинтетазы:
аспартат + глутамин + АТФ + Н2О → аспарагин + глутамат + АМФ + Н4Р2О7
Серии образуется их 3-фосфоглицерата - метаболита гликолиза.
Глицин образуется из серина под действием сериноксиметилтрансферазы:
серин + Н4-фолат → глицин + метилен- Н4-фолат + Н2О
Пролин синтезируется из глутамата:
Глутамат → у-полуальдегид глутамата → пролин
Цистеин образуется из серина и метионина. При этом метионин служит донором серы, а углеродный скелет и а-аминогруппа образуются из серина.
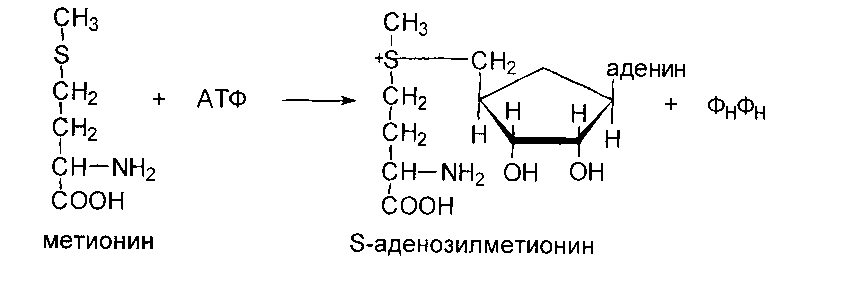
Обмен метионина и реакции трансметилирования
Метильная группа метионина - это мобильный одноуглеродный фрагмент, который используется для метилирования разных соединений. Непосредственным донором метальной группы в реакциях трансметилирования служит производное метионина - S-аденизилметионин, который образуется под действием метионин- аденозилтрансферазы из метионина и АТФ:
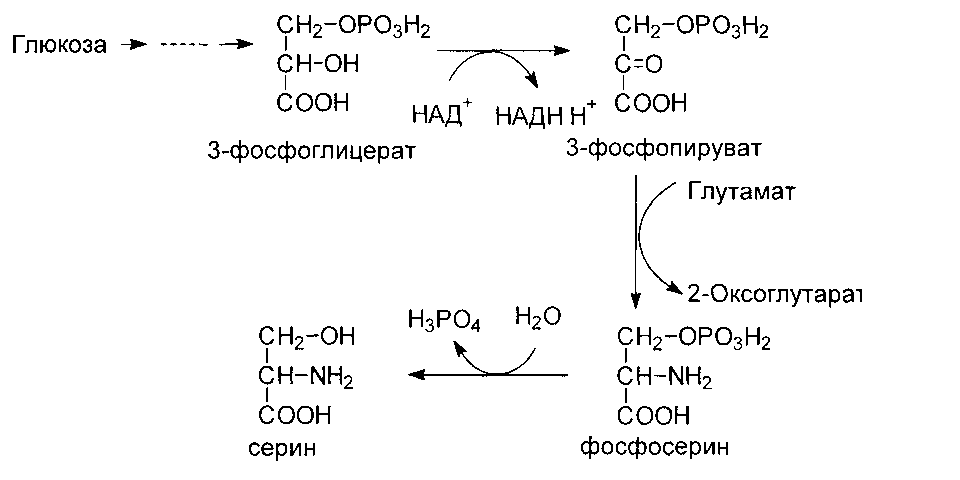
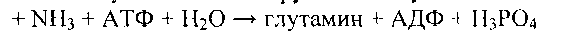
Непосредственно путем переаминирования метаболитов ОПК с глутаматом синтезируются аланин, аспартат и глутамат:
Пируват + глутамат → аланин + 2-оксоглутарат
Оксалоацетат + глутамат → аспартат + 2-оксоглутарат
2-оксоглутарат + аминокислота → глутамат + α-кетокислота
Глутамин синтезируется из глутамата под действием глутаминсинтетазы:
глутамат
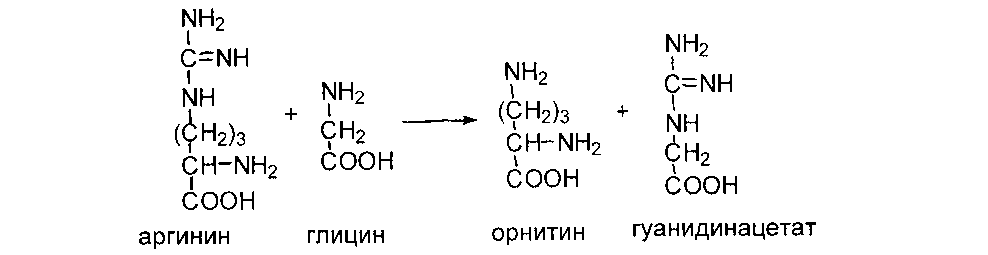
Рассмотрим реакцию трансметилирования на примере образование креатина - вещества, играющего важную роль в депонировании и транспорте энергии в мышечной ткани.
Сначала в почках образуется гуанидинуксусная кислота:
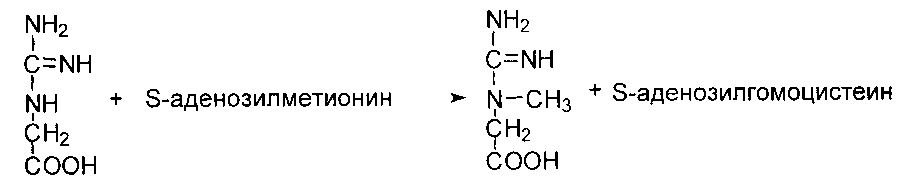
Далее гуанидинацетат транспортируется в печень, где в реакции трансметилирования превращается в креатин:
Обратимая реакция фосфорилирования креатина с образованием фосфокреатина катализируется креатинкиназой (КК).
Определение активности этого фермента и особенно его изофермента МВ-КК в сыворотке крови используется как тест повреждения сердечной мышцы.
Реакции метилирования принадлежит важная роль в образовании ряда биологически активных соединений (адреналина, фосфатидилхолина, креатина, карнитина и др.), а также в обезвреживании чужеродных соединений и биологически активных веществ (гистамина, катехоламинов и др.).
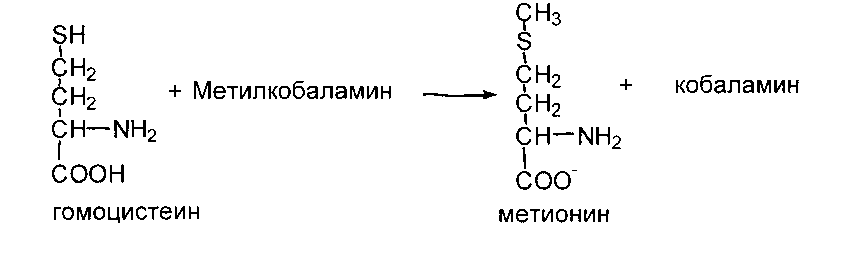
“Путь спасения” метионина
После отдачи метальной группы в реакции трансметилирования S- аденозилметионин (SAM) превращается в S-аденозилгомоцистеин (SAG). Под действием специфической гидролазы это вещество распадается на гомоцистеин и аденозин:
Гомоцистеин может вновь превращаться в метионин в реакции трансметилирования с 5-метил-Н4-фолатом:
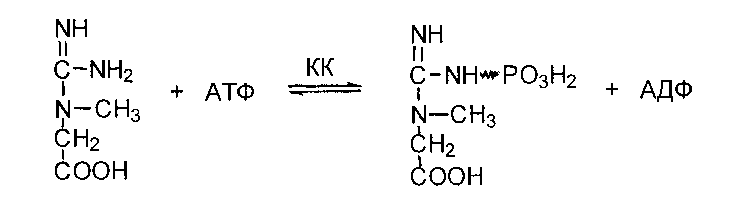
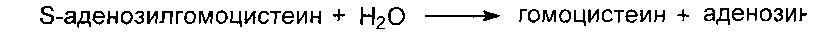
Реакция катализируется метионинсинтазой. Промежуточным переносчиком метальной группы в этой реакции служит метил-Н4-фолат - производное витамина В9. Таким образом, коферменты витаминов В12 и В9 тесно взаимодействуют в транспорте одноуглеродных радикалов. Поскольку гомоцистеин отсутствует в пище, данная реакция представляет собой “путь спасения” метионина.
Обмен одноуглеродных фрагментов
Для синтеза ряда соединений используются одноуглеродные фрагменты, такие как метальная группа (-СН3), метиленовая группа (-СН2-), метенильная группа (-СН=) и формильная группа (-СОН). Роль промежуточного переносчика этих групп играет Н4- фолат.
Н4-фолат образуется из фолиевой кислоты (фолата) при участии фолатредуктазы в печени. Коферментом фолатредуктазы является НАДФН-Н+.
Взаимопревращения производных тетрагидрофолата тесно связаны с обменом серина и глицина.
Превращение серина в глицин под действием серин-оксиметилтрансферазы происходит с участием Н4-фолата:
Обмен фенилаланина и тирозина
Фенилаланин - незаменимая аминокислота, т.к. в клетках животных не синтезируется бензольное кольцо.
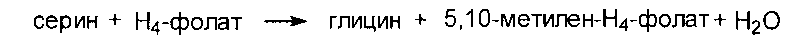
Распад глицина также происходит с участием Н4-фолата:
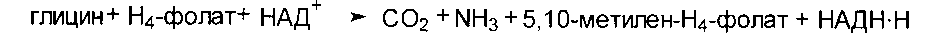
Метиленовая группа в молекуле 5,10-метилен-Н4-фолата может превращаться под действием специальных ферментов в другие одноуглеродные группы: 5,10-метенил-Н4-фолат и 10-формил-Н4-фолат. Все эти производные Н4-фолата служат донорами одноуглеродных радикалов при синтезе ряда соединений, в том числе дТМФ, пуриновых нуклеотидов, метионина и др.
Соответственно при гиповитаминозе, связанном с недостаточностью фолиевой кислоты, возникает дефицит предшественников ДНК и, в конечном счете, происходят изменения эритропоэза. Мегалобластическая анемия - почти всегда результат недостаточности фолиевой кислоты или витамина В12.
Фолиевая кислота является витамином не только для млекопитающих, но и для бактерий, в том числе болезнетворных. В последних фолиевая кислота образуется из парааминобензойной кислоты - одной из составных частей фолиевой кислоты. На структурном сходстве с парааминобензойной кислотой основано применение сульфаниламидных препаратов. При попадании в клетку бактерии сульфаниламидный препарат подавляет синтез фолиевой кислоты, нарушая все реакции, в которых она участвует. Размножение бактерий становится невозможным.
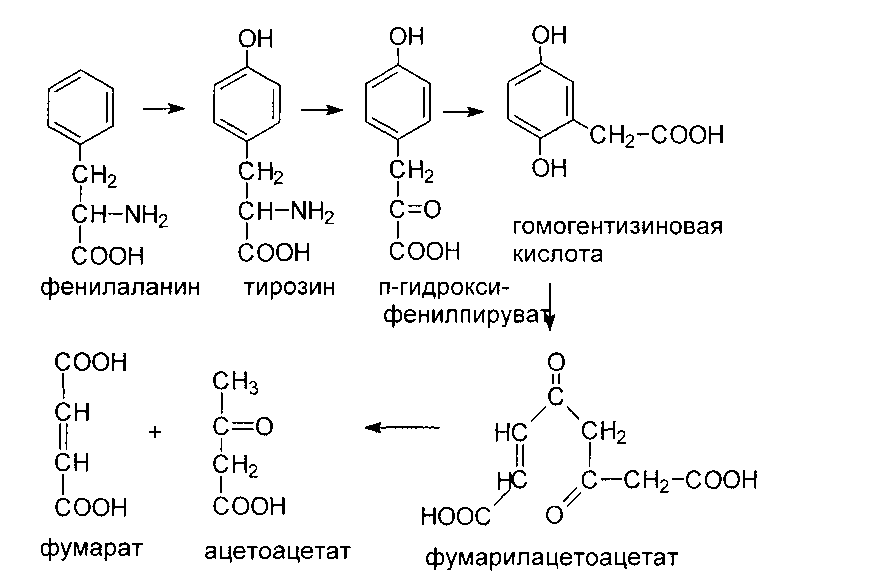
Основная масса фенилаланина утилизируется двумя путями - превращается в тирозин (90%) или включается в состав белков.
Превращение фенилаланина в тирозин катализируется ферментом фенилаланинмонооксигеназой, коферментом которой служит тетрагидробиоптерин (ТГБП). Для регенерации последнего используется НАДФН·Н+. Превращение фенилаланина в тирозин нужно скорее для удаления избытка фенилаланина, чем для образования тирозина, поскольку недостатка в тирозине обычно не бывает. При врожденном отсутствии этого фермента развивается заболевание фенилкетонурия.
Фенилкетонурия характеризуется нарушением обмена фенилаланина, в результате последний не может превращаться в тирозин и поэтому накапливается во всех жидкостях организма. Некоторые превращения фенилаланина, количественно несущественные у здорового человека, становятся заметными при фенилкетонурии. Наиболее значительным из них является переаминирование фенилаланина с образованием фенилпирувата. В основе самого названия болезни лежит высокое содержание этого фенилкетона в моче. Из фенилпирувата далее могут образовываться фениллактат, фенилацетат и О-гидроксифенилацетат.
Phe → фенилпируват → фенилацетат → фенилацетилглутамин
Конъюгат фенилацетата с глутамином выводится из организма с мочой.
Различают 2 формы фенилкетонурии: 1) классическая - наследственное заболевание связано с мутацией в гене фенилаланинмонооксигеназы. Наиболее тяжелые проявления - нарушение умственного и физического развития, судорожный синдром. 2) вариантная - следствие мутаций в генах, контролирующих метаболизм тетрагидробиоптерина. При этой форме клинические проявления близки, но не во всем совпадают с классической формой.
Нарушение умственного и физического развития при фенилкетонурии связано с токсическим действием на клетки мозга высоких концентраций фенилаланина, фенилпирувата и фениллактата. Большие концентрации Phe ограничивают транспорт Туг и Тгр через гематоэнцефалический барьер и тормозят синтез нейромедиаторов.
Более тяжелое течение вариантной формы фенилкетонурии связано с тем, что тетрагидробиоптерин необходим для реакций гидроксилирования не только Phe, но и Туг, и Тгр. Поэтому при недостатке этого кофермента нарушается метаболизм всех 3-х аминокислот, в том числе синтез нейромедиаторов - катехоламинов и серотонина. Заболевание характеризуется тяжелыми неврологическими нарушениями и ранней смертью (злокачественная фенилкетонурия).
При фенилкетонурии имеют место и другие нарушения аминокислотного обмена. Так, кожа и волосы у больных фенилкетонурией светлее, чем у их сибсов. Это обусловлено ингибированием реакции гидроксилирования тирозина - первого этапа в образовании пигмента меланина под влиянием высокой концентрации фенилаланина.
Лечение фенилкетонурии сводится к приему пищи с низким содержанием фенилаланина. Задача состоит в том, чтобы поступление фенилаланина в организм при данном заболевании не превышало потребности в нем для роста и замещения.
В связи с резко выраженной умственной отсталостью, развивающейся при фенилкетонурии, важное значение приобретает ранняя диагностика. С этой целью исследуют мочу новорожденного, добавляя в нее FeCl3. В присутствии фенилпирувата развивается оливково-зеленое окрашивание. Еще более надежным тестом считается определение фенилаланина в крови.
Частота встречаемости фенилкетонурии составляет 1 случай на 20000 новорожденных. Болезнь наследуется как аутосомный рецессивный признак. Гетерозиготы, составляющие
1,5% популяции, не обнаруживают видимых отклонений от нормы. Однако гетерозиготных носителей гена фенилкетонурии можно обнаружить с помощью теста толерантности к фенилаланину или по измерению кинетики исчезновения
внутривенно введенного фенилаланина. Эти тесты используются в генетической консультации для определения риска рождения больного ребенка.
Обмен тирозина значительно сложнее: тирозин используется для синтеза белков, служит предшественником катехоламинов, меланина, тироксина, а также может подвергаться катаболизму до СО2 и Н20.
Катаболизм тирозина. В результате ряда ферментативных превращений эти аминокислоты превращаются в фумарат и ацетоацетат.
Другим наследственным заболеванием, развивающимся как результат нарушения метаболизма тирозина, является алкаптонурия. Его непосредственной причиной является дефект фермента гомогентизат-диоксигеназы. В результате гомогентизат накапливается в жидкостях организма и выделяется с мочой, которая при стоянии чернеет, поскольку гомогентизат окислятся и полимеризуется в меланиноподобное соединение. Болезнь обычно обнаруживают по появлению черных пятен на пеленках. Других клинических проявлений и, прежде всего, нарушения умственного развития при данном заболевании не наблюдается.
Биосинтез меланина. В пигментных клетках из тирозина образуется пигмент меланин (от греч. melas - черный). При действии тирозинмонооксигеназы тирозин окислятся в дигидроксифенилаланин (ДОФА). ДОФА под действием тирозиназы - ключевого фермента всего процесса биосинтеза меланина превращается в ДОФА-хинон, из которого в результате неферментативных реакций образуется меланин. Меланин представляет собой группу полимерных соединений с неупорядоченной структурой. Цвет кожи и глаз зависит от количества и распределения меланоцитов и содержания в них меланина. Врожденное отсутствие тирозиназы в меланоцитах или отсутствие самих меланоцитов проявляется как альбинизм. Для этого заболевания характерны отсутствие пигментации кожи, волос и радужной оболочки глаз, сниженная острота зрения.
Обмен гистидина
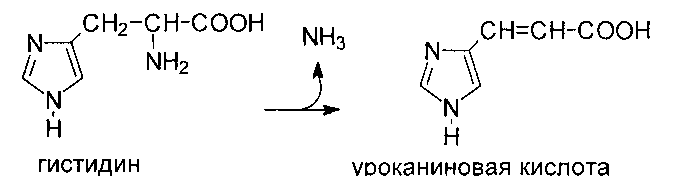
Катаболизм гистидина происходит путем его внутримолекулярного дезаминирования с образованием уроканиновой кислоты, которая затем через ряд реакций превращается в аммиак, одноуглеродный фрагмент, соединенный с тетрагидрофолатом, и
глутаминовую кислоту. Дезаминирование гистидина катализируется гистидин - аммиаклиазой, которая содержится в печени и коже:
И гистидин-аммиаклиазой, и уроканиназа появляются в крови при заболеваниях печени и измерение их активности используется для диагностики. Известна наследственная болезнь - гистидинемия, связанная с дефектом гистидин-аммиаклиазой. Для этого заболевания характерно повышенное содержание гистидина в тканях и нарушение физического и умственного развития.
Биогенные амины: их образование, функции и инактивация
Биогенные амины - органические соединения, которые образуются в результате декарбоксилирования аминокислот или их производных и имеют выраженную биологическую активность. В образовании биогенных аминов участвуют ферменты декарбоксилазы, коферментом которых служит пиридоксальфосфат.
Биогенные амины являются биологически активными веществами, которые выполнят функцию нейромедиаторов, гормонов, регуляторов местного действия.
Инактивация биогенных аминов происходит несколькими путями, основными из которых являются: 1) дезаминирование под действием ферментов моноаминоксидаз (МАО). Их коферментом служит ФАД. Сначала образуются альдегиды, а затем кислоты, которые выводятся почками.
R-CH2-NH2 → R-COH → R-COOH
2) путем метилирования под действием метилтрансфераз. При этом донором метальных групп является S-аденозилметионин (SAM).
Образование гистамина
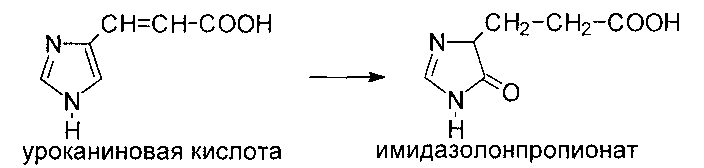
Уроканиновая кислота превращается в имидазолонпропионовую кислоту под действием уроканиназы, которая содержится только в печени:
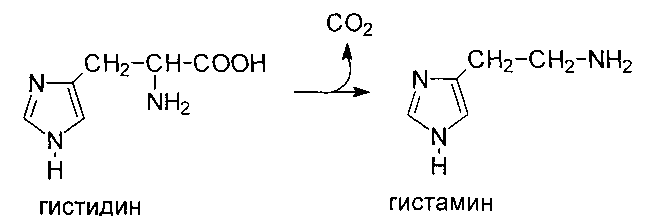
Декарбоксилирование гистидина катализируется гистидиндекарбоксилаза. Гистамин выполняет следующие функции: 1) участвует в развитии воспалительных реакций, 2) является медиатором аллергических реакций, 3) вырабатывается железами желудка и стимулирует секрецию желудочного сока.
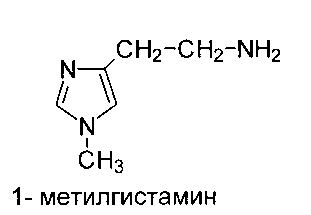
Инактивация гистамина происходит путем его метилирования с образованием 1- метилгистамина:
Образовавшийся 1-метилгистамин выводится из организма с мочой.
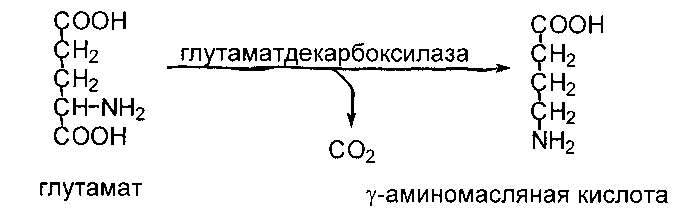
Образование 4-аминобутирата (ГАМК)
происходит в основном в пресинаптических отделах нейронов головного мозга. Наряду с глицином ГАМК является медиатором процесса торможения.
Инактивация ГАМК осуществляется путем переаминирования с 2-оксоглутаратом и последующим окислением образовавшегося сукцинат-полуальдегида в сукцинат:
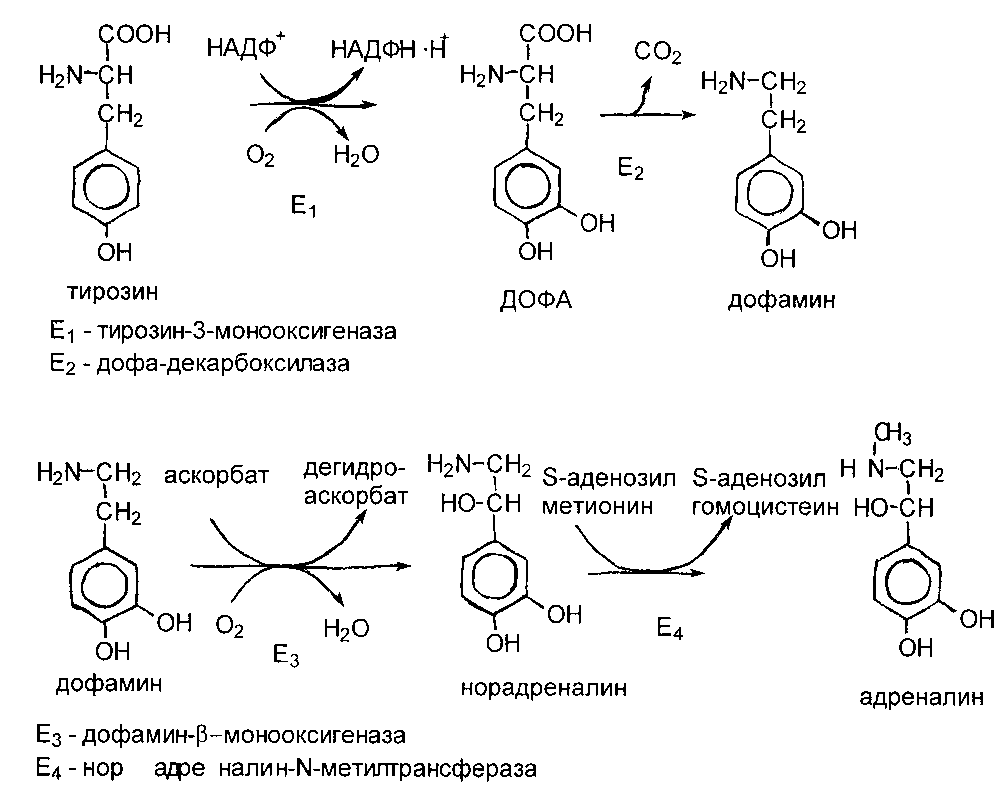
Образование катехоламинов
К важнейшим представителям катехоламинов относятся ДОФамин, норадреналин и адреналин. Все они образуются в мозговом веществе надпочечников и нервной ткани. Предшественником при синтезе служит тирозин:
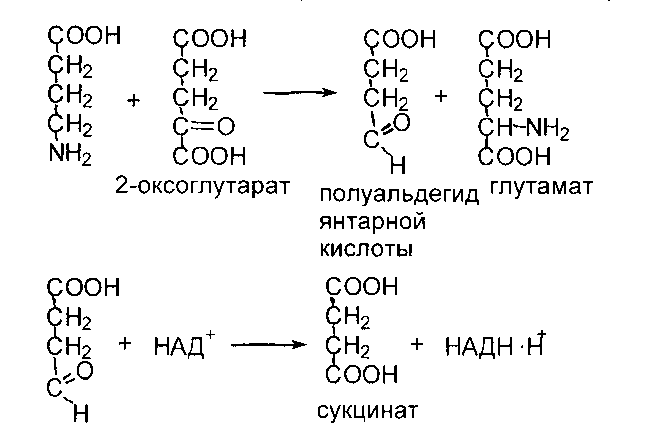
Катехоламины выполняют следующие функции: ДОФамин и норадреналин являются медиаторами в синаптической передаче нервного импульса; адреналин как гормон мозгового вещества надпочечников стимулирует мобилизацию депонированных углеводов и липидов, увеличивает силу и частоту сердечных сокращений и т.д.
Недостаточность дофамина в черной субстанции мозга приводит к болезни Паркинсона. Это одна из самых распространенных неврологических болезней - частота встречаемости 1 : 200 среди людей старше 60 лет. При этом заболевании снижена активность тирозинмонооксигеназы и ДОФА-декарбоксилазы. Заболевание сопровождается 3 основными симптомами: скованностью движений, ригидностью (напряжением) мышц и тремором (непроизвольное дрожание).
Инактивация катехоламинов происходит двумя путями: 1) в результате дезаминирования под действием фермента моноаминоксидазы (МАО), 2) метилирования под действием фермента катехол-О-метилтрансферазы (КОМТ).
Конечные продукты действия этих ферментов - метанефрины и ванилилминдальная кислота (ВМК) определяются в моче с целью диагностики феохромоцитомы - опухоли мозгового вещества надпочечников.
|
 Скачать 150.17 Kb.
Скачать 150.17 Kb.