|
|
гидрогеохимия. 1. введение Предмет и задачи гидрогеохимии, её место среди других наук, структура, основные этапы развития. Области применения гидрогеохимических данных
4. ФИЗИКО–ХИМИЧЕСКИЕ РАВНОВЕСИЯ В
ПОДЗЕМНОЙ ГИДРОСФЕРЕ
[Щелочно–кислотное равновесие, окислительно–восстановительное
равновесие, равновесие вода – минерал]
Мы начинаем рассмотрение физико–химических равновесий, которые или, вернее, приближение к которым существуют в подземной гидросфере. Можно говорить о равновесиях между молекулами Н2О и их ассоциатами, между ионами разнообразных химических элементов и молекулами и ионами воды; можно говорить о равновесии между различными химическими компонентами, растворёнными в воде, можно говорить о равновесии между водой и твёрдыми минералами, водой и породами, между водой и газом, между водой и органическим веществом, между водой и живым веществом. Всё это будут отдельные звенья того равновесия, к которому стремится сложнейшая система: подземная вода – горная порода – газ – органическое вещество. Одних из названных равновесий мы уже немного касались, других ещё коснёмся в дальнейших лекциях. А здесь рассмотрим три наиболее важных и наиболее изученных физико–химических равновесия: щелочно–кислотное, окислительно–восстановительное и равновесие вода – минерал.
Щелочно–кислотное равновесие. При обсуждении структуры воды мы говорили о том, что в воде присутствуют ассоциированные и мономерные молекулы. Теперь же обратим внимание на то, что в воде всегда присутствует и небольшое количество водородных (Н+) и гидроксильных (ОН–) ионов, поскольку вода как химическое соединение в малой степени диссоциирует по схеме H2O ↔ Н+ + ОН–. Строго говоря, ионы Н+ не существуют в воде в виде “чистого протона”, а образуют более сложные соединения типа гидроксония Н3O+. Но это принципиально не влияет на существо дела и можно условно принять указанную выше схему диссоциации. Произведение концентраций Н+ и ОН– называют ионным произведением воды. Независимые экспериментальные и расчётные исследования показали, что ионное произведение воды при температуре 22 °С равно 1·10–14. (Может возникнуть вопрос, в каких единицах измеряются концентрации Н+ и ОН–. Отвечаем, в молях на 1000 г воды. Такие единицы используются в термодинамике и они около цифр не проставляются). Если вода не содержит других ионов, то, исходя из требования электронейтральности, концентрации ионов Н+ и ОН– равны и каждая из них составляет 10–7.
Если от этой величины возьмём отрицательный десятичный логарифм, который обозначается буквой “р”, то получим уравнение рН = рОН = 7, характеризующее нейтральную реакцию раствора. Кислотно–щелочные условия раствора можно выражать с помощью величин рН и рОН, но принято это делать с помощью первого параметра. Если рН < 7, концентрация водородных ионов больше, чем гидроксильных — раствор кислый; если рН > 7, конценитрация водородных ионов меньше, чем гидроксильных — раствор щелочной.
При увеличении температуры диссоциация воды усиливается, и ионное произведение воды возрастает. Например, при 90 °С оно равно 10–12,4. Это означает, что при такой температуре рН чистой воды будет не 7, как при 22 °С, а 6,2.
Давление влияет на кислотно–щелочное состояние воды гораздо меньше, чем температура.
Важными факторами изменения щелочно–кислотных свойств подземных вод являются диссоциация кислот и оснований и гидролиз солей.
Способность кислот и оснований к отдаче ионов Н+ и ОН– характеризуется константами диссоциации этих кислот и оснований. По способности снижать рН кислоты располагаются в следующий ряд: НС1 > Н2SO4 > HF > H2CO3 > Н2S > H4SiO4.
По способности повышать рН подземных вод основания располагаются в такой ряд: NaOH > LiOH > Са(ОН)2 > Мg(ОН)2 > Fe(OH)2.
Горные породы в химическом отношении состоят в существенной степени из солей. Поэтому при взаимодействии пород с подземными водами широко распространён процесс гидролиза солей, влияющий на рН воды. Гидролиз (“разложение водой”) — это взаимодействие вещества с водой, при котором составные части вещества соединяются с составными частями воды. В зависимости от особенностей соли, подвергающейся гидролизу, влияние этого процесса на щелочно–кислотное состояние воды может быть разным. Рассмотрим наиболее типичные случаи.
1) Имеем соль слабой кислоты и сильного основания, например, соду Na2CO3. Её гидролиз сопровождается связыванием иона Н+ в слабодиссоциирующее соединение NаHCO3, в результате чего концентрация иона ОН– становится больше, чем концентрация иона Н+. Раствор даёт щелочную реакцию: СО32– + Н2О ↔ НСО3– + ОН–.
2) Имеем соль сильной кислоты и слабого основания, например, CuCl2. Ионы меди, соединяясь с гидроксильными ионами, образуют слабо диссоциирующие ионы СuОН+, при этом в растворе появляется избыток водородных ионов и раствор приобретает кислую реакцию: Сu2+ + Н2О ↔ СuОН+ + Н+.
3) В случае если гидролизу подвергается соль слабой кислоты и слабого основания, то процесс идёт в принципе так же, как в случаях 1 и 2, а реакция растворов будет зависеть от относительной силы соответствующих кислот и оснований и может быть нейтральной, слабокислой или слабощелочной.
4) Наконец, если имеем соль, образованную сильной кислотой и сильным основанием, например NaCl, то эта соль в воде полностью диссоциирует на ионы натрия и хлора. Ионы Na+ не будут связываться с ионами ОН–, а ионы С1– но будут связываться с ионами Н+, так как и NaOH, и НС1 полностью ионизируются. Таким образом, концентрации ионов Н+ и ОН– в растворе останутся неизменными, и раствор останется нейтральным. Другими словами, соли, образованные сильными кислотами и сильными основаниями, не подвергаются гидролизу.
С повышением температуры гидролиз солей интенсифицируется в связи с усилением при этом диссоциации воды и уменьшением констант диссоциации ряда кислот (H2CO3, H2S и других) и оснований (Са(ОН)2, Мg(ОН)2).
Значения рН подземных вод в большинстве случаев составляет 6–8,5. Однако могут быть значительно выше и значительно ниже. Они изменяются от < 0 до 12,5. В соответствии с классификацией подземных вод по кислотности–щелочности, предложенной В.В. Ивановым и Г.А. Невраевым, выделяются воды: сильнокислые (рН < 3,5), кислые (3,5–5,5), слабокислые (5,5–6,8), нейтральные (6,8–7,2), слабощелочные (7,2–8,5) и щелочные (> 8,5).
Минимальными значениями рН (< 0 2) характеризуются термальные воды и конденсаты вулканических газов районов современного магматизма. Кислые воды (рН < 2 5) распространены в зонах окисления сульфидных месторождений. Кислая реакция среды (рН 4–5 и менее) характерна для высококонцентрированных хлоридных кальциевых рассолов. Сильнощелочная реакция свойственна содовым подземным водам, а также рудничным водам, связанным с разложением нефелиновых сиенитов.
Щелочно–кислотные условия в значительной степени влияют на миграцию химических элементов. У многих из них имеются определённые кислотно–щелочные диапазоны увеличения и уменьшения концентраций в подземных водах. Наиболее сильно щелочно–кислотные свойства подземных вод влияют на миграцию так называемых элементов–гидролизатов, к которым относятся: А13+, Со3+, Cr3+, Bi3+, Sn2+, Th4+, Zr4+, Ti4+, Sc3+ и др. Эти элементы склонны образовывать гидроокиси, причём при довольно низких значениях рН. Например, гидроокись А1 начинает выпадать при рН около 4. Поэтому А1 и другие элементы–гидролизаты мигрируют и образуют высокие концентрации только в весьма кислых водах.
Другие элементы, например, такие как Na+, К+, Rb+, Сs+, Sr2+ не образуют гидроокислов в земной коре, и для их миграции и осаждения рН вод не имеет существенного значения.
Некоторые элементы (Аs, Мо, Gе, Sе) имеют кислую и щелочную области увеличения концентраций в подземных водам. В кислых водам их миграция осуществляется в виде растворимых кислот (Н3АsО4, Н2МоО4 и др.), а в щелочных — в виде анионов этих кислот, образующих хорошо растворимые соединения с Na.
Окислительно–восстановительное равновесие. Рассмотрим химическую реакцию, которая может происходить в грунтовой воде болотным ландшафтов:
8e
8Fe3+ + Н2S + 4Н2О ↔ 8Fe2+ + SO42– + 10Н+.
Взаимодействуют ион трёхвалентного железа с сероводородом, образующимся при разложении органического вещества. В результате образуются ионы двухвалентного железа и сульфата. Это окислительно–восстановительная реакция. Произошло восстановление трёхвалентного железа до двухвалентного, и произошло окисление сульфидной серы с валентностью –2 до сульфатной с валентностью +6. Но что такое изменение валентности? Это или потеря, или приобретение электронов. В данном случае атом (ион) серы потерял восемь электронов, а каждый из восьми ионов железа приобрёл по одному электрону (итого восемь).
Таким образом, сущность окисления заключается в потере электронов атомами или ионами окисляющего вещества, а сущность восстановления — в присоединении электронов к атомам или ионам восстанавливающегося вещества. Окисление какого–либо вещества не может произойти без одновременного восстановления другого вещества, так как свободные электроны не могут накапливаться в растворе (раствор должен быть электронейтральным). В рассмотренной нами реакции железо является окислителем, сероводород — восстановителем.
Подземные воды содержат большое число элементов с переменной валентностью. Эти элементы участвуют в окислительно–восстановительных реакциях. К таким элементам относятся: Fe (+2, +3), Mn (+2, +3, +4), U (+4, +6), S (–2, +2, +4, +6), Сu (+1, +2), Се (+3, +4), Р (+3, +5), V (+3, +4, +5), Cr (+3, +6) и другие.
Важнейшим окислителем является кислород. К важным окислителям относятся S (в форме SO42–), С (СО2), Fe (Fe3+), Mn (Мn4+, Мn3+). Важнейшими восстановителями являются молекулярный водород (Н2), сероводород (Н2S), соединения углерода (СН4, различные органические соединения), Fe (Fe2+) и др.
Каждая совокупность разновалентных соединений (ионов и молекул) одного какого–либо элемента является окислительно–восстановительной системой.
Надо отметить, что большая часть элементов, хотя и имеет переменную валентность, но из–за небольших концентраций не определяет характер окислительно–восстановительного состояния подземных вод. Эти элементы лишь, так сказать, подстраиваются под те окислительно–восстановительные условия, которые определяются широко распространёнными элементами с переменной валентностью. Эти определяющие элементы образуют в подземным водах системы, которые называются потенциалзадающими. Это системы серы, железа, кислорода, органических веществ, водорода.
Существование в подземных водах окислительно–восстановительных систем отдельных элементов приводит к установлению в них динамического равновесия, характеризующего окислительно–восстановительное состояние подземной воды в целом. Окислительно–восстановительное состояние подземной воды определяет характер окислительно–восстановительной среды или обстановки.
С целью количественной оценки окислительной и восстановительной способности вод используют показатель Eh, который называется окислительно–восстановительным (или редокс–) потенциалом и измеряется в вольтах (или милливольтах). Поскольку сущность окислительно–восстановительных реакций состоит в перемещении электронов, то окислительно–восстановительный потенциал (Eh) представляет собой меру электронного обмена данной реакции по сравнению со стандартной, за которую принята реакция окисления водорода.
Важно понимать, что для каждого элемента окислительная и восстановительная среда характеризуются различными величинами Eh. Например, при Eh = +0,7 В среда может являться восстановительной для трёхвалентного железа, но окислительной для двухвалентной меди. Кроме того, природные окислительно–восстановительные процессы протекают при участии ионов водорода, и ход этих процессов зависит от рН среды, увеличение которого приводит к уменьшению Eh окислительно–восстановительной системы. Поэтому в геохимии активно пользуются диаграммами pH – Eh, а разделение обстановок по Eh на окислительные и восстановительные имеет смысл, строго говоря, только применительно к определённым элементам.
Несмотря на это, существующие наиболее общие представления об окислительно–восстановительных обстановках подземных вод таковы.
Окислительные обстановки характеризуются присутствием в водах свободного кислорода или других сильных окислителей. Железо, марганец, медь, ванадий, сера находятся в высоких степенях окисления (Fe3+, Мn4+, Сu2+, S6+). Главный критерий окислительной обстановки — присутствие свободного кислорода в водах. Если свободный кислород отсутствует, то показателем окислительных обстановок является трёхвалентное железо.
Главным критерием восстановительных обстановок служит двухвалентное состояние железа и отсутствие свободного кислорода. Среди восстановительных обстановок можно различать сероводородную и бессероводородную (или глеевую). В первой в водах присутствуют H2S, железо и многие другие металлы обычно немиграционноспособны, образуют труднорастворимые сульфиды. В восстановительной бессероводородной обстановке важное место занимают метан, прочим углеводороды, двухвалентное железо. Многие металлы здесь легко мигрируют, т.е. хорошо растворимы.
Сравнение восстановительной бессероводородной и восстановительной сероводородной обстановок показывает, что геохимические процессы зависят не столько oт Eh (в обеих они могут быть одинаковыми), сколько от природы восстановителя. В присутствии H2S, например, Fe2+ не мигрирует; в его отсутствие в восстановительных условиях — мигрирует хорошо.
По современным данным пределы Eh подземных вод ограничены цифрами –600 +860 мВ. Eh подземных вод обычно уменьшается с ростом рН. Максимальные значения Eh (+860 мВ) обнаружены в кислых водах (рН < 2), а минимальные (–600 мВ) — в резкощелочных (рН
12,5).
Различные геохимические типы подземных вод имеют свои пределы изменений окислительно–восстановительного потенциала. Так, в кислородсодержащих водах Еh, как правило, более 200 мВ. Высокие значения Eh имеют кислые воды окисляющихся сульфидных месторождений (до +600 и даже +860 мВ). Низкие величины Eh обнаружены в минерализованных водам и рассолах; взаимодействующих с нефтяными залежами и битуминозными породами, и в водах, содержащих сероводород; их Eh опускается до –400 –500 мВ.
Действие различных потенциалзадающих систем подземных вод (и пород), приводит к тому, что в геологических структурах формируется окислительно–восстановительная зональность подземных вод, которая выражается в закономерном изменении Eh этих вод при их движении по геологической структуре, что имеет существенные минералогические последствия. Наиболее яркий пример окислитально–восстановительной зональности — это так называемые инфильтрационно–эпигенетические месторождения урана.
На крыле артезианского бассейна имеется песчаный водоносный горизонт, перекрытый и подстилаемый глинистыми водоупорами, В водоносный горизонт из области питания, где находятся кристаллические породы с несколько повышенной концентрацией урана, фильтруются подземные воды. Звёздочками показана часть пласта, которая подверглась пластовому окислению. Здесь залегают лимонитизированные окраснённые пески. Правая часть горизонта — восстановительная обстановка: серые пески с включениями пирита. На контакте окислительной и восстановительной зон имеет место восстановительный геохимический барьер. Здесь хорошо растворимый шестивалентный уран восстанавливается до четырёхвалентного и выпадает в виде UO2, создавая оруденение.
Геохимическое равновесие вода – минерал. Поскольку подземные воды всегда находятся в горных породах, а породы состоят из минералов, то исключительно важно определить, способна ли данная вода растворять тот или другой минерал, или она может его высаживать. Вообще говоря, возможны три случая: 1) вода находится в равновесии с минералом; она не способна ни растворять, ни осаждать его; 2) вода не насыщена по минералу, она способна растворять его; 3) вода перенасыщена по минералу, она способна высаживать его. Оценка того, какой из трёх случаев имеет место, осуществляется с помощью довольно сложных расчётов, которые можно выполнить несколькими способами на основании положений классической, то есть равновесной термодинамики. Надо сказать, что в свете современных представлений природные процессы неравновесны, или необратимы, и должны описываться с помощью не классической, а неравновесной термодинамики. Однако аппарат последней ещё недостаточно разработан. И применение равновесной термодинамики для описания природных процессов оправдывается так называемым принципом частичного, или мозаичного, или локального равновесия, который предполагает, что неравновесный (необратимый) в целом процесс может быть разбит на ряд элементарных пространственно–временных этапов, в каждом из которых выполняются условия термодинамического равновесия.
Итак, введём первое понятие, которое нам необходимо для оценки равновесности системы вода – минерал — произведение растворимости минерала. Если в стакан с дистиллированной водой (t = 18 °С) погрузить кусочек минерала, например, гипса (СаSO4 · 2Н2О), он начнёт растворяться. В воду начнут переходить ионы Са2+ и SO42–. Рано или поздно наступит момент, когда растворение прекратится. В этот момент количество ионов Са2+ и SO42–, “отрываемых” водой от кристаллической решётки минерала, будет равно количеству ионов Са2+ и SO42–, вовлекаемых в кристаллическую решётку обратно из воды. Это момент термодинамического равновесия: раствор и минерал находятся в равновесии. Если в этот момент измерить концентрации Са2+ и SO42– в растворе, выразить их, как принято в термодинамике, в молях на литр (т.е. разделить концентрацию в г/л на атомную или ионную массу) и перемножить эти концентрации, то мы получим величину 10–4,44. Это и есть произведение растворимости (ПР) гипса. Запишем это в виде уравнения:
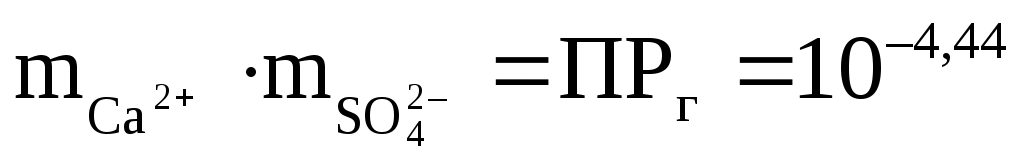
(m — означает молярное выражение концентраций).
Для кальцита (СаСО3) уравнение будет выглядеть так:
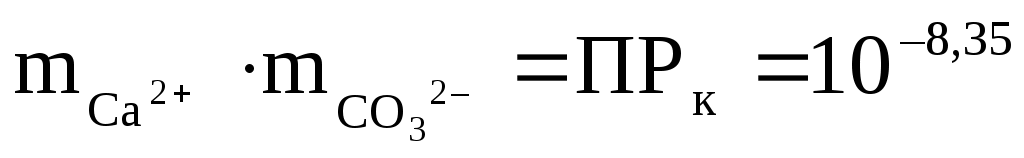 . .
Для доломита [CaMg(CO3)2], где в молекуле два иона СО32–, уравнение пишется так:
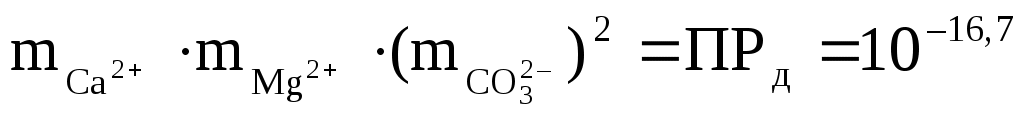
(Количество одинаковых ионов в молекуле анализируемого минерала в соответствии с законом действующих масс, входит в уравнение в виде степени).
Величина ПР указывает на растворимость минерала: чем больше ПР, тем больше растворимость. У гипса она больше, чем у кальцита, а у кальцита больше, чем у доломита. Значения ПР для очень многих веществ определены экспериментально и приводятся в химических и термодинамических справочниках.
Можем ли мы произвести аналогичный расчёт для природного раствора, чтобы узнать, насколько близко состояние этого раствора к состоянию равновесия с тем или иным минералом? Можем в том случае, если уясним, в чём заключается разница между сложным природным раствором и, так сказать, идеальным раствором, который использовался для введения понятия ПР и который не содержал никаких других компонентов, кроме тех ионов, которые образуют кристаллическую решётку минерала. Специфика природного раствора состоит в двух основных моментах.
1) В природном растворе кроме интересующих нас ионов есть много других. Например, если мы хотим оценить состояние природного раствора по отношению к гипсу, то нас, прежде всего, интересуют Ca2+ и SO42–. А в растворе, кроме того, всегда присутствуют Mg2+, Na+, Cl–, HCO3–, CO32– и др. Присутствие этих последних мешает Ca2+ и SO42– эффективно взаимодействовать. Ведь, что такое, допустим, кристаллизация гипса из раствора? Смысл её в том, что Ca2+ и SO42– должны сблизиться настолько, чтобы взаимное притяжение стало больше разъединяющих их сил молекул воды. А если в эти отношения вмешаются другие ионы, то ситуация усложняется. Поэтому для реальных растворов вместо концентрации компонентов используется величина активности, которая является эффективной концентрацией или количеством вещества, реально участвующим в данной реакции, и которая представляет собой произведение концентрации иона (m) на его коэффициент активности (γ). Например, 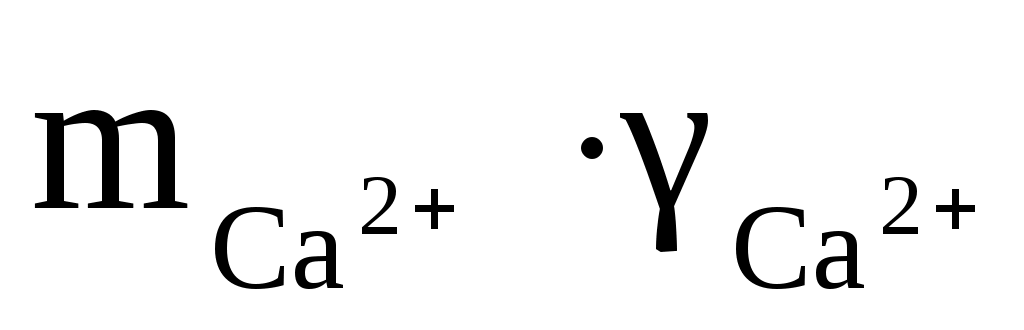 или или 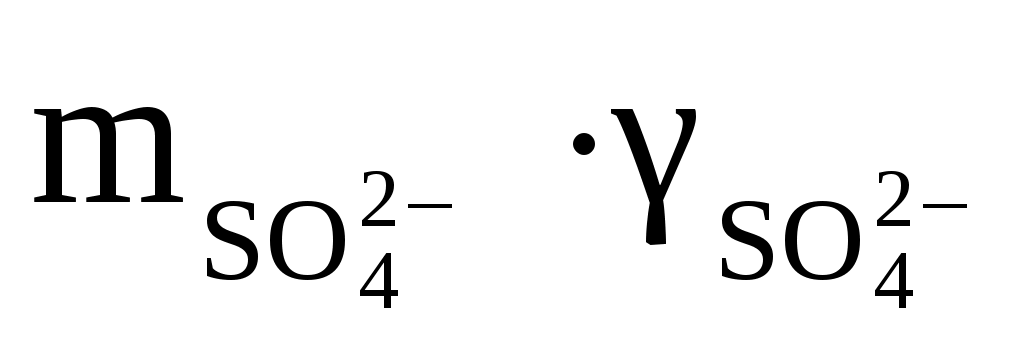 (В том “идеальном” растворе, который мы обсуждали при определении понятия ПР, активность компонентов равна их концентрации). (В том “идеальном” растворе, который мы обсуждали при определении понятия ПР, активность компонентов равна их концентрации).
2) В природном растворе всегда в том или ином количестве присутствуют не только свободные ионы, которые реально участвуют в реакциях, но и более сложно построенные частицы, которые называются комплексными ионами и ионными парами. Например, в гидрокарбонатной кальциевой воде кроме Ca2+, HCO3–, CO32–, Mg2+, Na+, SO42–, Cl– присутствуют CaCO30, CaHCO3+, CaSO40, MgCO30, MgHCO3+, MgSO40, NaCO3–, NaSO4– и др. Одним словом, свободных ионов в растворе меньше, чем устанавливается химическим анализом. Нами для конкретных проб подземных вод района Паричей, например, рассчитано, что при М 171 мг/л доля свободных ионов от их аналитической концентрации составляет для Ca2+ 96,5 %, для Mg2+ 97,0 % и для CO32– 43,1 %; при М 1046 мг/л эти показатели соответственно 92,4, 92,7 и 27,0 %. С дальнейшим ростом минерализации доля свободных ионов ещё больше снижается.
Таким образом, уравнения для реальных растворов, аналогичные тем, что мы писали для “идеальных” растворов при определении ПР, будут иметь следующий вид. Для гипса:
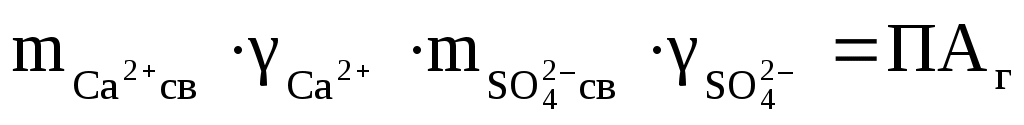 , ,
для кальцита:
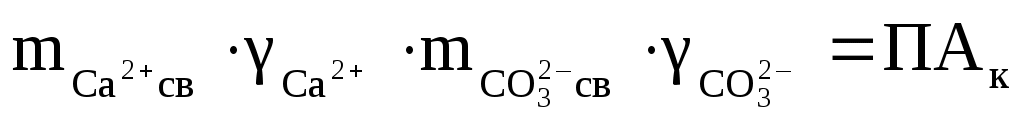 , ,
для доломита:
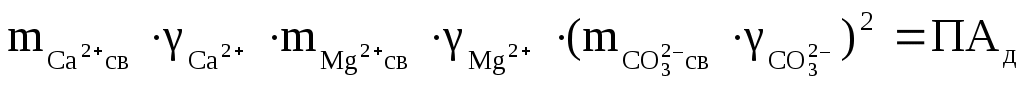 , ,
где: 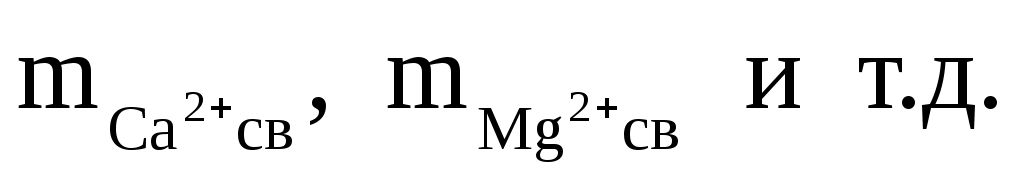 — моляльность соответствующих ионов, не связанных в комплексы. — моляльность соответствующих ионов, не связанных в комплексы.
Надо обратить внимание на то, что в правой части уравнений стоит величина ПА. Это произведение активностей — аналог величины произведения растворимости (ПР) для природных растворов. Мы уже почти подошли к определению степени равновесности природного раствора. Но прежде, чем это сделать, мы должны рассмотреть, как определяется концентрация свободным ионов и как определяются коэффициенты активности ионов.
Надо сказать, что наибольшую сложность при подобных термодинамических оценках составляет определение доли свободных ионов. Это делается путём совместного решения уравнений баланса масс и диссоциации комплексных ионов, которое сводится к решению систем уравнений такого вида:
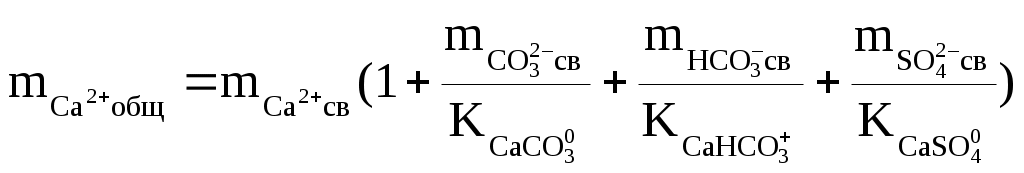
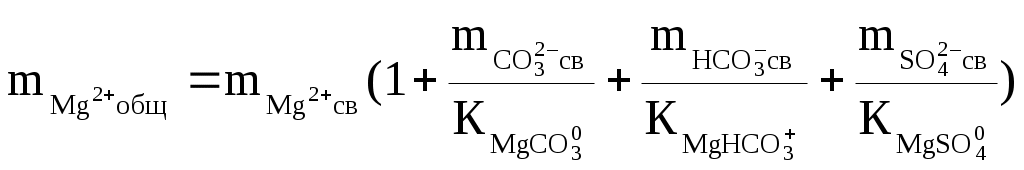
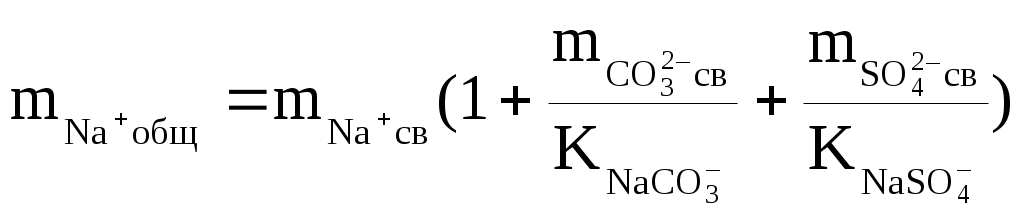
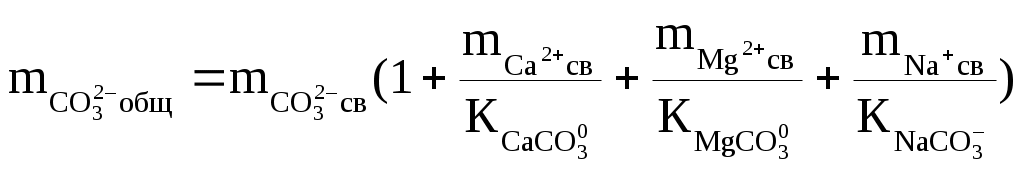
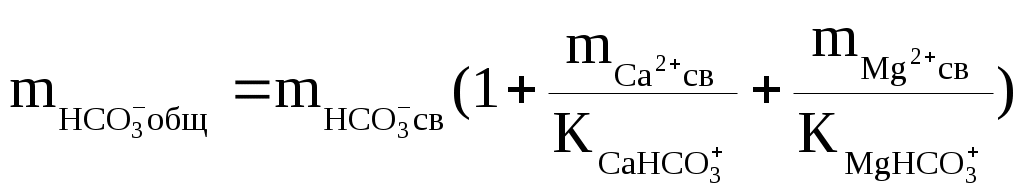
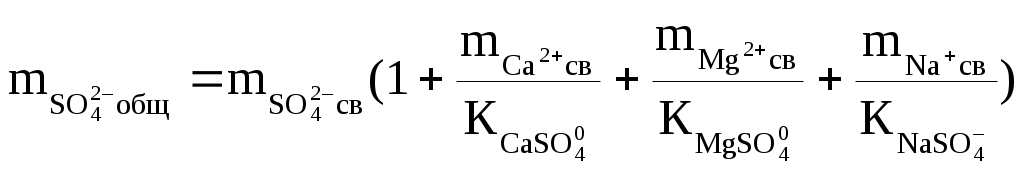 , ,
где: 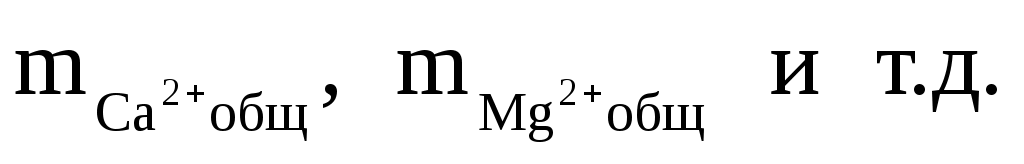 — общие (аналитические) моляльности соответствующих ионов; — общие (аналитические) моляльности соответствующих ионов; 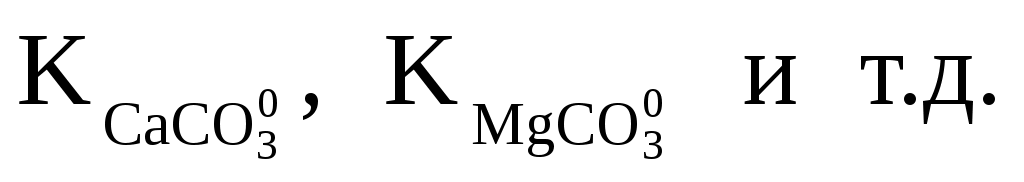 — концентрационные константы диссоциации соответствующих комплексов. — концентрационные константы диссоциации соответствующих комплексов.
Такие системы уравнений решаются на ЭВМ методом последовательного приближения.
Коэффициенты активности индивидуального иона (i) вычисляются по формуле Дебая–Хюккеля:
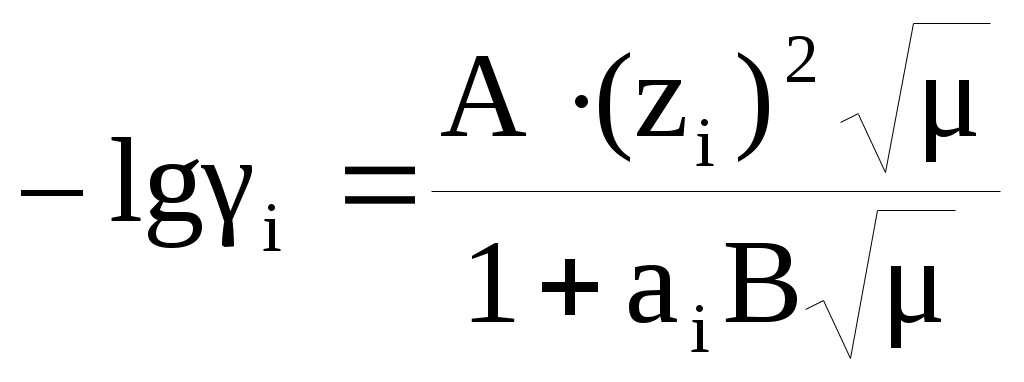 , ,
где: А, В — особые константы растворителя, зависящие от t и Р и табулированные в справочниках; аi — множитель, зависящий от “эффективного диаметра” иона i (даётся в справочниках); zi — заряд иона i в данном растворе; — ионная сила.
Ионная сила — это параметр, который сильно связан с минерализацией раствора, но в отличие от неё позволяющий учесть электростатическое взаимодействие разноимённых ионов в растворе. Она определяется по следующей формуле:
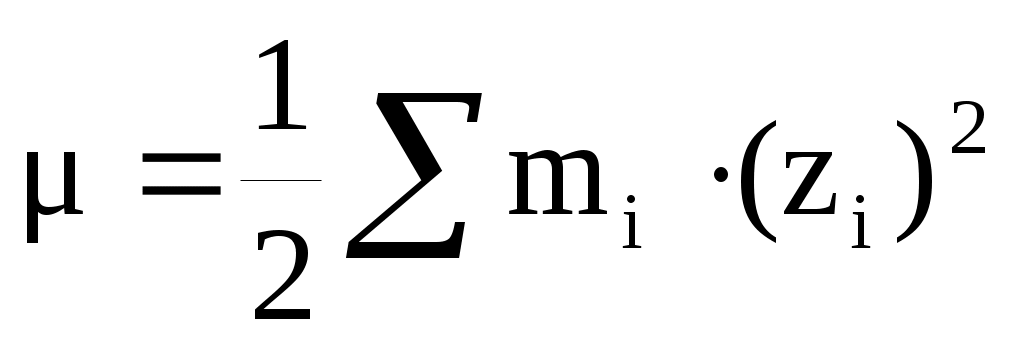 , ,
где: mi — моляльность, zi — заряд иона i в данном растворе,
Наконец, мы подошли к цели нашим расчётов и можем рассмотреть, как оценивается степень равновесности реального раствора с данным минералом, или, что то же, степень насыщенности раствора по отношению к данному минералу. Обозначим её буквой А.
Степень насыщенности (А) равна логарифму отношения произведения активностей (ПА) к произведению растворимости (ПР):
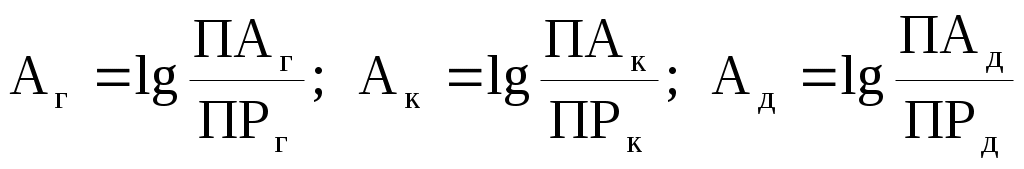 . .
Если величина А равна нулю, раствор находится в равновесии с минералом и не может ни растворять, ни осаждать его. Если А < 0, раствор не насыщен по данному минералу и может растворять его. Если А > 0, раствор перенасыщен по данному минералу и может осаждать его. Перенасыщенность раствора означает, что при прохождении раствором равновесного состояния кристаллизация вещества не началась в силу каких–то причин (например, из–за отсутствия центров кристаллизации).
Важно заметить, что конечный результат может быть дан как в относительных единицах, так и в массовых. В последнем случае можно сказать, сколько, например, граммов того или иного минерала может раствориться в 1 литре данной воды (или какая масса минерала может выпать из 1 литра воды).
Надо сказать, что расчёты по схеме, которую мы рассмотрели, можно осуществлять для пресных, солоноватых и солёных растворов с ионной силой не более 0,7, которая соответствует морской воде. Для растворов с более высокой минерализацией, когда количество взаимодействующих между собой простых и комплексных ионов существенно возрастает, применяются другие, более сложные, способы расчётов.
5. ФОРМИРОВАНИЕ ХИМИЧЕСКОГО (КОМПОНЕНТНОГО) СОСТАВА
|
|
|
 Скачать 395 Kb.
Скачать 395 Kb.