Аналитика (1-14 вопросы). Дисциплина Аналитическая химия и фхма
 Скачать 0.55 Mb. Скачать 0.55 Mb.
|

|
Важнейшие окислительно-восстановительные индикаторы
Редокс-индикаторы должны удовлетворять определенным требованиям: – индикатор должен быть чувствительным, т.е. реагировать в точке эквивалентности с минимальным количеством окислителя или восстановителя; – окраска окисленной и восстановленной форм должна резко отличаться; – изменение окраски должно быть отчетливо заметно при применении небольшого количества индикатора; – интервал перехода индикатора должен быть невелик, и совпадать со скачком титрования; – индикатор должен быть устойчив по отношению к кислороду воздуха, углекислому газу, свету и т.п. Иногда в окислительно-восстановительном титровании используют так называемые необратимые индикаторы. В конечной точке титрования они необратимо окисляются избытком окислителя и при этом меняют свою окраску. К необратимым индикаторам относятся метиловый оранжевый, метиловый красный, конго красный и др. Например, метиловый оранжевый окисляется по схеме: (CH3)2NC6H4N  NC6H4SO3Na → (CH3)2NC6H4NO + ONC6H4SO3Na NC6H4SO3Na → (CH3)2NC6H4NO + ONC6H4SO3Na12. Перманганатометрия: сущность метода, общая характеристика, область применения. Перманганатометрический метод анализа основан на реакциях окисления восстановителей перманганатом калия. Перманганат калия является одним из сильнейших окислителей. В зависимости от концентрации ионов водорода в растворе, восстановление перманганат-иона протекает различным образом. В сильнокислой среде МnO4− восстанавливается до Мn2+: МnO4− + 8Н+ + 5е Мn2+ + 4Н2О (2.1) Е0(MnO4–/Mn2+) = + 1,51 В, В слабощелочной, нейтральной или слабокислой среде МnO4− восстанавливается до МnO2: МnO4− + 2Н2О + 3е МnO2 + 4ОН– (2.2) Е0(MnO4–/MnО2) = + 0,57 В В анализе некоторых органических веществ используется титрование в сильнощелочной среде: МnO4− – 1е МnO42− (2.3) Е0(MnO4–/ MnO42–) = + 0,56 В Перманганатометрия широко используется для определения: - восстановителей: Fe2+, Sn2+, SO32−, S2O32−, NO2−, S2−, I−, Br−, различных органических соединений и др. (обычно используется метод прямого титрования): 5Fe2+ + MnO4– + 8H+ = 5Fe3+ + Mn2+ + 4H2O (2.4) Если восстановитель с перманганатом взаимодействует медленно, применяют обратное титрование. Для этого к раствору восстановителя приливают точно измеренный объем перманганата калия, заведомо взятого в избытке; после завершения окислительно-восстановительной реакции избыток не вступившего в реакцию перманганата оттитровывают стандартным раствором другого восстановителя (щавелевая кислота, оксалаты, соли железа (II) и т.п.) - окислителей: Fe3+, NO3−, ClO3−, Cr2O72−, H2O2 и др. (метод обратного титрования). Окислители восстанавливают титрованным раствором восстановителя и избыток последнего оттитровывают перманганатом калия. Например, к раствору К2Cr2O7 (окислителя) добавляют избыток соли Мора (восстановителя), бихромат восстанавливается по уравнению: 6 Fe2+ + Cr2O72− + 14 H+ → 6 Fe3+ + 2 Сr3+ + 7 H2O Избыток соли Мора, не вступившей в реакцию, титруют раствором перманганата по реакции (2.4). - веществ, не проявляющих окислительно-восстановительных свойств, но способных реагировать с окислителями или восстановителями: Ca2+, Sr2+, Ba2+, Pb2+, Zn2+ и др. (метод обратного титрования или метод замещения). Например, при определении кальция в анализируемый раствор, содержащий ионы Ca2+, добавляют вспомогательный реагент – оксалат натрия или аммония; при этом образуется эквивалентное количество осадка оксалата кальция: Ca2+ + C2O42– → СаC2O4↓ В методе обратного титрования оксалат берут заведомо в избытке, осадок обычно отделяют, избыток оксалата титруют перманганатом: 5 C2O42− + 2 MnO4− + 10 H+ → 10 CO2 + 2 Mn2+ + 8 H2O (2.5) При использовании метода замещения осадок оксалата кальция отфильтровывают, промывают водой и растворяют на фильтре серной кислотой: CaC2O4 + H2SO4 → СаSO4 + H2C2O4 Выделяется эквивалентное количество щавелевой кислоты, которую титруют раствором перманганата по реакции (2.5). Обычно в перманганатометрии применяют титрование в кислой среде т.к. в этом случае окислительная способность перманганата калия значительно выше. Кроме того, в кислой среде образуются бесцветные ионы Мn2+, а в нейтральной и щелочной средах – бурый осадок МnO2, который затрудняет фиксирование точки эквивалентности. Для создания среды при определении концентрации титруемых растворов чаще всего применяют серную кислоту, т. к. азотная кислота сама является сильным окислителем и может способствовать протеканию нежелательных побочных процессов, а соляная кислота окисляется перманганатом калия до свободного хлора. Однако, многие вещества (особенно органические) определяют в нейтральной и щелочной средах, т. к. они количественно окисляются только в присутствии щелочи, а в кислых растворах такие соединения окисляются лишь частично (например, сульфиты, сульфиды, тиосульфаты, гидразин и др.). При определении органических соединений используется восстановление перманганата по схеме (2.3). Органические соединения при этом обычно окисляются до карбоната. По окончании реакции восстановления перманганата в щелочной среде раствор подкисляют и титруют избыток MnO4− раствором подходящего восстановителя. Например, метанол в щелочной среде окисляется перманганатом по реакции: CH3OH + 6 MnO4− + 8 OH− → CO32− + 6 MnO42− + 6 H2O Для осаждения манганат-иона в раствор заранее вводят BaCl2 . Этим методом можно определить также муравьиную, винную, лимонную, салициловую и другие кислоты, глицерин, фенол, формальдегид и другие органические соединения. При титровании перманганатом в большинстве случаев не требуется индикатор, т.к. даже очень разбавленные растворы его интенсивно окрашены, и одна лишняя капля KMnO4 окрашивает раствор в розовый цвет, появление которого свидетельствует о завершении реакции титрования. По сравнению с другими методами окислительно-восстановительного титрования рассматриваемый метод отличается рядом достоинств. 1. Т.к. одна капля раствора KMnO4 даже при концентрации его, равной 0,01 н., окрашивает в конце титрования 50 мл раствора в отчетливый розовый цвет, титрование можно проводить, не прибегая к использованию индикаторов по появлению розовой окраски при прямом титровании или по её исчезновению при обратном. 2. Титрование перманганатом можно осуществлять в кислой и щелочной средах. 3. Перманганат имеет очень высокий окислительно-восстановительный потенциал (в кислой среде Е0(MnO4–/Mn2+) = + 1,51 В). Поэтому многие вещества, которые невозможно оттитровать более слабыми окислителями, можно определять методом перманганатометрии. 4. Перманганат является дешевым и легкодоступным реагентом. 5. Перманганат можно применять для определения веществ, не обладающих окислительно-восстановительными свойствами (см. выше). Перманганатометрический метод имеет и некоторые недостатки. 1. Перманганат калия трудно получить в химически чистом виде, поэтому точную концентрацию приготовленного раствора необходимо устанавливать путем стандартизации. 2. Растворы перманганата со временем меняют свой титр, поэтому в процессе использования их необходимо периодически проверять. 3. Титрование не следует проводить в солянокислых растворах, т.к. перманганат окисляет ионы Cl− до свободного хлора. 4. Некоторые реакции окисления перманганатом протекают при комнатной температуре очень медленно, поэтому иногда требуется нагревание растворов. Приготовление раствора перманганата калия. Основным рабочим раствором метода является 0,1 н. или 0,05 н. раствор KMnO4. Перманганат калия реактивной квалификации (ч.д.а. – чистый для анализа или х.ч. – химически чистый) всегда содержит примеси продуктов восстановления МnO2, следовые количества иона Мn2+. При растворении перманганата калия в воде в присутствии Мn2+ происходит реакция: 2 МnO4− + 3 Мn2+ + 2 Н2О 5 МnO2 + 4 Н+, но даже в отсутствии Мn2+ реакция разложения перманганата калия катализируется твердым диоксидом марганца. Разложение KMnO4 протекает по уравнению: 4 МnO4− + 2 Н2О 4 МnO2 + 3 O2 + 4 ОН–. Кроме того, органические вещества, находящиеся в дистиллированной воде или на поверхности сосуда для хранения, восстанавливают перманганат-ион до МnO2 . Вследствие этого концентрация раствора KMnO4 в первое время после приготовления несколько уменьшается. Отсюда следует, что приготовить титрованный раствор перманганата по точной навеске нельзя. Обычно свежеприготовленный раствор перманганата выдерживают 7-10 дней (или кипятят в течение 1-2 часов) для завершения окислительно-восстановительных процессов. По истечении этого времени выделившийся осадок МnO2 отфильтровывают через невосстанавливающийся фильтр (пористый стеклянный или воронку со стеклянной ватой) или сливают раствор при помощи сифона. Приготовленный раствор хранят в темных бутылях со стеклянными пробками. Навеска перманганата калия для приготовления раствора рассчитывается по формуле: 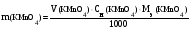 Из уравнения МnO4− + 8 Н+ + 5е Мn2+ + 4 Н2О следует, что 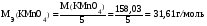 . .Точную концентрацию приготовленного раствора перманганата калия устанавливают путем стандартизации. Наиболее удобными стандартными веществами являются щавелевая кислота H2C2O4 ˙ 2H2O и ее соли – оксалаты, т.к. они легко могут быть очищены от примесей перекристаллизацией из раствора. Стандартизация раствора перманганата калия по щавелевой кислоте Реакция окисления щавелевой кислоты перманганатом калия выражается суммарным уравнением 5 H2C2O4 + 2 KMnO4 + 3 H2SO4 = 2 MnSO4 + 10 CO2 + K2SO4 + 8 H2O (3.4) C2O42− – 2e → 2 CO2 ↓ 5 MnO4− + 8 H+ +5e → Mn2+ + 4 H2O ↑ 2 В действительности процесс протекает значительно сложнее, с образованием промежуточных соединений и для его начала необходимо присутствие в растворе хотя бы следов Mn2+. Анионы MnO4− медленно реагируют с ионами щавелевой кислоты непосредственно, но быстро – с ионами Mn2+: MnO4− + Mn2+ + C2O42– → MnO42− + MnC2O4+ Манганат-ион MnO42− в кислом растворе быстро диспропорционирует: Mn (VI) + Mn (II) = 2 Mn (IV) Mn (IV) + Mn (II) = 2 Mn (III) Марганец (III) образует оксалатные комплексы состава Mn(C2O4)n(3−2n)+, где n = 1, 2, 3; они медленно разлагаются с образованием Mn2+ и CO2. Таким образом, реакция (3.4) является автокаталитической, катализатором которой служат ионы Mn2+. Пока в растворе присутствует незначительное количество Mn2+, реакция протекает очень медленно. Когда же концентрация Mn2+ достигает определенной величины, реакция начинает протекать с большой скоростью. Сложный характер процесса обуславливает ряд особенностей при титровании щавелевой кислоты перманганатом калия. Установлено, что наиболее воспроизводимые и точные результаты получаются, если около 90 % необходимого количества перманганата добавлять при комнатной температуре и слабом перемешивании, затем повысить температуру до 70-80 0С и закончить титрование при 55-60 0С. 13.Йодометрия: сущность метода, общая характеристика, область применения. Условия проведения йодометрических определений. Йодометрия - метод окислительно-восстановительного титрования, основанный на реакциях, связанных с окислением восстановителей свободным йодом I2 или с восстановлением окислителей йодидом калия KI. Оба процесса можно выразить следующей схемой: I2 + 2е 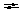 2 I− (2.8) 2 I− (2.8)Т.к. стандартный окислительно-восстановительный потенциал пары невелик − E0(I2 /2I−) = 0,54 В, йод является относительно слабым окислителем, а ионы I− – сравнительно сильным восстановителем. С помощью метода йодометрии можно определять: 1. Восстановители SO32−, S2O32−, NO2−, S2−, CN−, SCN− и др.: а) путем прямого титрования анализируемого раствора раствором йода: SO32− + I2 + 2Н2О = SO42− + 2 I− + 2H+ ; (2.9) б) путем обратного титрования, Используется, если скорость взаимодействия восстановителя с йодом невелика. В этом случае к раствору восстановителя добавляют избыток титрованного раствора I2, а спустя некоторое время не вступивший в реакцию йод титруют раствором тиосульфата натрия: 2 S2O32− + I2 = S4O62- + 2 I− (2.10) 2. Окислители Fe3+, Cr2O72-, H2O2, Cl2, Br2, ClO3−, MnO4− и др. Проводить определение, титруя раствор окислителя йодидом калия KI нельзя, т.к. невозможно фиксировать конец титрования. Поэтому пользуются методом замещения: к анализируемому раствору окислителя добавляют избыток раствора KI: 2 MnO4− + 10 I− + 16 H+ = 5 I2 + 2 Mn2+ + 8 Н2О. (2.11) В результате реакции выделяется йод в количестве, эквивалентном количеству окислителя; йод титруют раствором тиосульфата натрия по уравнению (2.10). К достоинствам метода йодометрии можно отнести следующие: 1. Большая точность по сравнению с другими методами окислительно-восстановительного титрования. 2. Растворы йода окрашены, и титрование можно проводить без индикатора. Желтая окраска ионов I3− при отсутствии других окрашенных продуктов отчетливо видна при очень малой концентрации (5∙10−5н.). 3. Йод хорошо растворяется в органических растворителях, поэтому метод широко применяется для титрования в неводных средах. Недостатки метода, вызывающие ошибки при выполнении йодометрических определений: 1. Потери йода из-за его летучести. Поэтому титрование проводят на холоду и по возможности быстро. При необходимости оставить раствор на некоторое время для завершения реакции, его хранят под притертой пробкой. 2. Окисление ионов йода кислородом воздуха в кислой среде. 3. Йодометрическое титрование нельзя проводить в щелочной среде вследствие диспропорционирования йода. 4. Относительно медленные скорости реакций с участием йода. 5. В процессе хранения стандартные растворы йода и тиосульфата изменяют свой титр. Чтобы избежать ошибок, необходимо периодически проверять титр йода по тиосульфату, а титр тиосульфата по дихромату калия. Фиксируют конечную точку титрования в методе йодометрии с помощью специфического индикатора – крахмала, который образует с йодом комплексно-адсорбционное соединение синего цвета. Эта реакция очень чувствительна, она позволяет легко обнаруживать йод при концентрации 10−5 н. Т.к. соединение йода с крахмалом очень прочное, крахмал следует добавлять в конце титрования, когда окраска раствора становится бледно-желтой. Если вводить крахмал раньше, то очень много йода будет связано с крахмалом. При титровании йод с трудом освобождается из соединения с крахмалом, и результат титрования окажется неточным. При проведении йодометрических определений следует соблюдать следующие условия: 1. Т.к. стандартный окислительно-восстановительный потенциал пары I2/2I− невелик, многие йодометрические определения не доходят до конца. Поэтому для количественного протекания реакций необходимо создавать специальные условия (вводить комплексообразователи, осадители, буферные добавки и т.д.). 2. Йод – вещество летучее, поэтому титрование проводят на холоду. Кроме того, при увеличении температуры снижается чувствительность крахмала как индикатора (при 50 0С индикатор в 10 раз менее чувствителен, чем при 25 0С). 3. Растворимость йода в воде мала, поэтому определение окислителей необходимо проводить в присутствии большого избытка KI, который образует с йодом растворимое нестойкое комплексное соединение: KI + I2 K[I3] (2.12)4. Скорость реакции между окислителями и KI обычно невелика, поэтому к титрованию выделившегося йода обычно приступают спустя некоторое время. 5. Йодометрическое титрование нельзя проводить в щелочной среде, т.к. протекает побочная реакция: I2 + 2 OH− = IO− + I− + Н2О (2.13) Образующийся гипойодит является более сильным окислителем, чем йод, он окисляет тиосульфат до сульфата: S2O32− + 4 IO− + 2 OH− = 4I− + 2SO42− + Н2О. Поэтому во избежание побочных реакций титрование проводят при рН не более 9. 6. В кислых растворах йодиды постепенно окисляются кислородом воздуха: 4I− + O2 + 4H+ = 2I2 + 2 Н2О (2.14) Свет ускоряет эту реакцию, поэтому реакционную смесь хранят в темноте. Рабочими растворами метода йодометрии являются растворы йода и тиосульфата натрия. Титрованный раствор йода можно приготовить исходя из точной навески химически чистого кристаллического йода, который очищают от примесей путем возгонки. Однако, очистка йода представляет собой очень трудоемкую операцию. Кроме того, титрованный раствор в процессе работы с ним и при длительном хранении меняет свой титр вследствие летучести йода, и периодически его нужно проверять. Поэтому обычно готовят раствор I2 приблизительно нужной концентрации (0,05–0,1 н.) растворением навески йода в растворе KI (40 г/л). Точную концентрацию полученного раствора устанавливают по раствору тиосульфата натрия (реакция 2.10). Тиосульфат натрия Na2S2O3·5Н2О является неустойчивым веществом. Оно легко реагирует с углекислым газом и кислородом воздуха: Na2S2O3 + Н2О + СO2 = NaHCO3 + NaНSO3 + S↓ 2Na2S2O3 + O2 = 2Na2SO4 + 2S↓ Поэтому готовят приблизительно 0,1 н. раствор тиосульфата, растворяя навеску соли в свежепрокипяченой воде (для удаления СO2). Хранить готовый раствор Na2S2O3 рекомендуется в темных бутылях, защищенных от двуокиси углерода трубкой с натронной известью. В дальнейшем титр раствора начинает медленно уменьшаться, поэтому его необходимо периодически проверять. Для установки концентрации тиосульфата предложено много различных стандартных веществ, например твердый химически чистый йод, йодат калия KIO3, бромат калия KBrO3, дихромат калия и др. На практике чаще всего пользуются дихроматом калия K2Cr2O7. В качестве стандартного в-ва используют K2Cr2O7, но т.к он с тиосульфатом взаимодействует не стехеометрично используют метод замещения. К подкисленному раствору KJ добавляют раствор дихромата и спустя 5-10 мин выделившийся йод титрую тиосульфатом по уравнению K2Cr2 O7+6KJ+JH2SO4=Cr2(SO4)3+3J2+4K2SO4+7H2O 2 S2O32− + I2 = S4O62- + 2 I− | ||||||||||||||||||||||