Реферат неврология. Хорея Гентингтона. Тип наследования, клиника, диагностика, лечение, профилактика.
 Скачать 492.07 Kb. Скачать 492.07 Kb.
|

|
Приднестровский государственный университет имени Т. Г. Шевченко Медицинский факультет Кафедра травматологии, ортопедии и экстремальной медицины РЕФЕРАТ По дисциплине Неврология, медицинская генетика, нейрохирургия На тему: «Хорея Гентингтона. Тип наследования, клиника, диагностика, лечение, профилактика.» Выполнила: Студентка 4 курса 407 АКП Специальности «Лечебное дело» Станкова Е. В. Проверила: Ассистент Окушко Светлана Владимировна г. Тирасполь, 2021г. Оглавление1.Краткая информация 2 Определение 3 Этиология и патогенез 3 Кодирование по МКБ-10 5 Классификация 5 2. Клинические проявления. 6 3. Диагностика и дифференциальный диагноз. 8 4.Лечение 9 5. Профилактика 11 Список использованных источников 12 1.Краткая информацияОпределениеБолезнь Гентингтона (БГ) — это наследственное неуклонно прогрессирующее нейродегенеративное заболевание с аутосомно-доминантным типом наследования и полной пенетрантностью, развивающееся вследствие экспансии тринуклеотидных повторов CAG в первом экзоне гена HTT, локализованного на коротком плече четвёртой хромосомы и кодирующего белок гентингтин. Аутосомно-доминантный тип наследования означает, что (1) лица обоего пола с одинаковой вероятностью могут носить мутацию БГ; (2) БГ может передаваться как по мужской, так и по женской линии; (3) каждый ребёнок в семье с родителем, являющимся гетерозиготным носителем мутации БГ, имеет 50 %-ный риск унаследования мутации этого заболевания (в случае гомозиготного носительства мутации БГ последняя передастся каждому представителю потомства). В настоящее время не существует одобренных методов, позволяющих излечить, замедлить или остановить прогрессирование БГ, однако должное симптоматическое лечение пациентов с этим заболеванием позволяет улучшить качество жизни как самих пациентов, так и ухаживающих за ними лиц и родственников 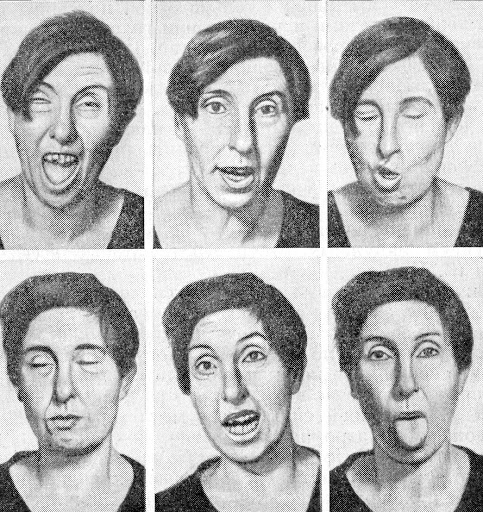 Этиология и патогенезБГ развивается вследствие мутации в гене HTT (ранее известном как IT-15), который расположен в дистальном участке короткого плеча четвёртой хромосомы (область 4p16.3). Суть мутации заключается в экспансии (увеличении количества) тринуклеотидных повторов CAG (цитозин–аденин–гуанин) в первом экзоне указанного гена более 35. Таким образом, наличие у субъекта CAG-повторов в количестве 36 и более даже на одном из аллелей гена HTT будет означать носительство мутации БГ. Это приводит к тому, что с мутантного гена синтезируется белок гентингтин с патологической структурой — избыточным содержанием остатков аминокислоты глутамина. Крайне редко встречаются случаи гомозиготного носительства мутации БГ. Примечательно, что гомозиготное носительство мутации не влияет значимым образом на клинические особенности и возраст дебюта БГ по сравнению с гетерозиготным носительством. Экспрессия гена HTT наблюдается во всех органах и тканях, однако в наибольшей степени она выражена в головном мозге и яичках. Функция белка гентингтина остаётся до конца не выясненной, однако известно, что гентингтин, содержащий патологический полиглутаминовый фрагмент, токсичен для клеток головного мозга и приводит к их прогрессирующей гибели. С патоморфологической точки зрения, наиболее характерным для БГ является постепенно нарастающая атрофия структур полосатого тела (хвостатое ядро и скорлупа) с последующим вовлечением в патологический процесс других областей головного мозга, включая кору больших полушарий. Примечательно, что, по данным наблюдательных исследований PREDICT–HD и TRACK–HD, незначительно выраженные клинические проявления моторной дисфункции, когнитивных и поведенческих изменений начинают проявляться у носителей мутации БГ ещё до момента (примерно, за 10 лет) установления клинического диагноза этого заболевания, а избирательная региональная атрофия вещества головного мозга начинает развиваться уже за 12–15 лет до клинического дебюта БГ. БГ характеризуется полной пенетрантностью, т.е. при наличии соответствующей мутации заболевание развивается в 100 % случаев, если субъект доживает до соответствующего возраста. Возраст дебюта БГ имеет обратно пропорциональную зависимость от количества CAG-повторов — чем их больше, тем раньше появляются симптомы заболевания. Так, при наличии CAG-экспансии в 36–39 повторов дебют БГ с большой вероятностью можно ожидать после 65 лет, а в случае 60 CAGповторов и более вероятнее всего развитие ювенильной формы БГ с дебютом в возрасте 20 лет и ранее. На долю ювенильной формы БГ, в среднем, приходится около 5 % пациентов. Таким образом, учитывая, что носитель мутации может не дожить до возраста дебюта БГ в силу иных причин, формально, наличие CAG-экспансии в 36–39 повторов характеризуется неполной пенетрантностью. Вместе с тем, величина CAG-экспансии определяет возраст дебюта БГ примерно на 56 % — остающаяся вариабельность может быть следствием как влияния иных генетических различий (в том числе мозаицизм и соматическая нестабильность CAG-повторов), так и действия факторов внешней среды. Таким образом, БГ может дебютировать в любом возрасте — преимущественно, в зависимости от величины CAG-экспансии — и характеризуется наличием у субъектов периода асимптомного носительства мутации. Величина экспансии CAG-повторов, как правило, нарастает в последующих поколениях, особенно при передаче мутантного гена по отцовской линии — так называемый феномен «отцовской передачи». Увеличение из поколения в поколение числа CAGповторов приводит к более раннему проявлению в каждом последующем поколении симптомов БГ (феномен антиципации). Кодирование по МКБ-10G10 - Болезнь Гентингтона (включено: хорея Гентингтона) КлассификацияПринимая во внимание особенности генетики БГ, носительство мутации этого заболевания может быть разделено на асимптомный (premanifest) и симптомный (manifest) периоды. Асимптомный период подразделяется на досимптомный (presymptomatic), в течение которого у носителя мутации БГ не отмечается никаких субъективных, клинических или измеряемых инструментально изменений (как правило, это 10–15 лет до дебюта БГ), и продромальный (prodromal), который характеризуется постепенным появлением слабо выраженных двигательных, когнитивных и (или) поведенческих изменений, не позволяющих при этом поставить формально клинический диагноз БГ. Существует несколько вариантов классификации симптомных носителей мутации БГ. По стадиям заболевания в зависимости от оценки по шкале общей функциональной способности (Total functional capacity, или TFC), являющейся разделом Унифицированной шкалы оценки болезни Гентингтона (Uniňed Huntington’s Disease Rating Scale, или UHDRS), БГ имеет следующую классификацию:  По клиническому фенотипу на основании превалирования определённого синдрома условно различают следующие формы БГ: 1. «Классическая» гиперкинетическая. 2. Ригидная: ювенильная (вариант Вестфаля) — дебют в 20 лет и раньше; поздняя: нередко является следствием прогрессирования БГ с трансформацией клинической картины гиперкинетического синдрома в брадикинезию с экстрапирамидной мышечной ригидностью; примерно у 10 % взрослых пациентов дебют БГ происходит с акинетико-ригидного синдрома с минимальными проявлениями хореи. 3. Психическая (не имеет самостоятельного значения и выделяется при резком превалировании психических симптомов над неврологической симптоматикой). 2. Клинические проявления.Наиболее характерными проявлениями являются хореический гиперкинез и прогрессирующая деменция. Классическая гиперкинетическая форма у большинства пациентов дебютирует в возрасте 30 лет и старше. Задолго до появления развернутой клинической картины постепенно развивается неусидчивость, повышенная активность пациентов, движения становятся размашистыми, нередко – нескоординированными, становится излишне оживленной жестикуляция. Хореический гиперкинез характеризуется быстрыми, неритмичными, беспорядочными движениями в различных мышечных группах с вовлечением мускулатуры конечностей, лица, диафрагмы, туловища. Обращают на себя внимание гримасничанье больных, нескоординированные, размашистые движения в руках, неустойчивость, пошатывание при ходьбе. Выполнение произвольных движений затруднено вследствие гиперкинезов и сопровождается рядом ненужных движений. Так, при ходьбе больной может раскачиваться из стороны в сторону, приседать, приплясывать. Гиперкинезы могут провоцироваться выполнением целенаправленных действий (кинезиогенные гиперкинезы), эмоциональным напряжением, волнением. При разговоре появляются или усиливаются избыточные движения губ, языка, других мышц, что проявляется причмокиванием, облизыванием губ языком, покашливанием, шмыганьем носом. Отличительной особенностью болезни Гентингтона является то, что на протяжении некоторого времени, особенно в начале заболевания, пациент способен сознательно подавлять гиперкинезы. Характерным является отсутствие гиперкинезов глазодвигательной мускулатуры. По мере прогрессирования заболевания в патологические движения вовлекаются все более обширные мышечные группы. Гиперкинезы приобретают большую амплитуду, в меньшей степени поддаются произвольному контролю. Помимо хореического гиперкинеза возникают атетоидные движения в пальцах кистей рук, мышечная дистония в конечностях и туловище. Вследствие вовлечения в процесс аксиальной мускулатуры (мышцы туловища), резко затрудняется походка, возникает неустойчивость при ходьбе, стоянии. Поражение бульбарной мускулатуры может приводить к выраженной дизартрии и дисфагии. Тяжелые расстройства глотания могут явиться причиной возникновения аспирационной пневмонии. Характерными являются нарушения психики и когнитивные расстройства. Нередко заболевание дебютирует именно поведенческими нарушениями. В начальных стадиях заболевания преобладают депрессивные, тревожные расстройства. Характерными являются неусидчивость, нарушения внимания, снижение памяти и ограничение способности к усвоению нового материала, повышенная возбудимость. У отдельных больных могут наблюдаться более сложные нарушения поведения в виде склонности к бродяжничеству (дромомания), бредовое толкование окружающего, галлюцинации. Когнитивные нарушения характеризуются относительно медленной прогредиентностью, благодаря чему больные могут определенное время сохранять работоспособность в рамках привычной деятельности. В случае прогрессирования заболевания когнитивные нарушения могут достигать степени тяжелой деменции. Заболевание постепенно прогрессирует, своего максимума гиперкинезы достигают через 5-10 лет. В последующем, мышечная гипотония и гиперкинезы могут сменяться мышечной ригидностью и акинезией. Примерно у 10% больных на поздних стадиях заболевания развивается полная обездвиженность и контрактуры. Выраженные гиперкинезы и тяжелое слабоумие (деменция) являются основными причинами инвалидизации. Ведущими причинами наступления летального исхода являются присоединяющиеся инфекционные осложнения (аспирационная пневмония, сепсис вследствие инфекции мочевыводящих путей). Среди пациентов с выраженными депрессивными расстройствами высока частота суицидальных попыток. Ювенильная форма Вестфаля. Наблюдается примерно в 5-10 % всех случаев болезни Гентингтона. Характеризуется более ранним началом (второе десятилетие жизни). В отличие от классической формы заболевания, экстрапирамидные нарушения проявляются ранним возникновением акинетико-ригидного синдрома (повышение мышечного тонуса по пластическому типу, замедленность движений – брадикинезия, ограничение спектра спонтанных движений – олигокинезия). Одновременно у больных развиваются и быстро прогрессируют признаки поражения больших полушарий (эпилептические припадки, деменция) и мозжечка (атаксия, интенционный тремор). Особенностью данной формы является выраженная и быстро прогрессирующая деменция. Первыми проявлениями могут быть интеллектуальные расстройства, в дальнейшем постепенно развивается деменция. Прогноз ювенильной формы болезни Гентингтона более тяжелый, заболевание быстро инвалидизирует больного, летальный исход наступает через 8-10 лет после появления клинических проявлений.  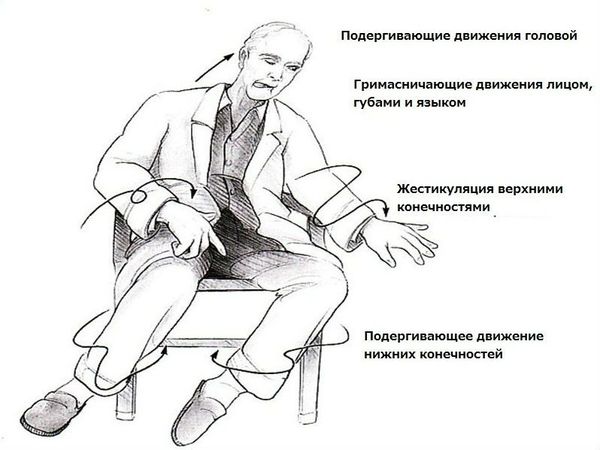 3. Диагностика и дифференциальный диагноз.К критериям диагностики болезни Гентингтона относят: дебют классической формы заболевания в 30-40 лет, акинетико-ригидной - в 10-15 лет; сочетание экстрапирамидных нарушений и деменции; аутосомно-доминантный тип наследования с практически полной пенетрантностью мутантного гена; экспансия тринуклеотидных повторов ЦАГ свыше 36 в генеIT-15 (выявляется при прямой ДНК - диагностике). Наиболее точным диагностическим критерием болезни Гентингтона является молекулярная диагностика, обеспечивающая возможность не только выявления мутантного гена, но и точного установления числа ЦАГ - повторов, что позволяет прогнозировать тяжесть и темп прогрессирования заболевания. Возможна пренатальная диагностика на основании анализа клеток из амниотического мешка. Важно, что диагностика не требует обследования нескольких членов семьи. Методы нейровизуализации (МРТ) проводятся для дифференциальной диагностики болезни Гентингтона с заболеваниями, имеющими сходные клинические проявления, но характеризующимися специфическими радиологическими признаками (например, рассеянный склероз). Дифференциальная диагностика проводится с другими видами хореических гиперкинезов, в основе которых лежат различные этиологические факторы. Малая хорея (хорея Сиденгама). Возникает в детском или подростковом возрасте. Развитие малой хореи связано с перенесенной стрептококковой инфекцией. В основе заболевания лежит выработка антител к антигену стрептококка, способных перекрестно реагировать с антигенами нейронов подкорковых ядер. Течение заболевания характеризуется наличием периодов ухудшения состояния (атаки), продолжающихся 3 - 6 месяцев. Когнитивные и психические нарушения, возникающие одновременно с двигательными расстройствами, могут сохраняться на протяжении длительного периода времени, в том числе, и после исчезновения гиперкинезов. Диагноз устанавливается на основании связи проявлений хореи с перенесенной инфекций, клинической картиной ревматизма. Лечение включает санацию очага инфекции, проведение антибактериальной терапии (при необходимости – с использованием плазмафереза, кортикостероидов), симптоматической терапии. Прогноз заболевания благоприятный, у значительного числа пациентов наступает полное выздоровление. Нейроакантозитоз-хорея. Редкое генетически детерминированное заболевание, проявляющееся сочетанием хореического гиперкинеза и аномалией строения эритроцитов. Наследуется по аутосомно-рецессивному типу. Считалось эндемичным для южного и восточного Средиземноморья, однако, в последние годы появились сообщения о выявлении заболевания и в других регионах, в частности, среди коренного населения Якутии. При этом заболевании значительное число эритроцитов приобретает неправильную (рогообразную, полулунную) форму, изменяются биохимические свойства их клеточных мембран. Неврологическая симптоматика характеризуется сочетанием хореического гиперкинеза, реже – акинетико-ригидного синдрома и аксональной полинейропатии (угнетение сухожильных и периостальных рефлексов, мышечные гипотрофии в дистальных отделах конечностей). Патогенетической терапии не существует, лечение носит симптоматический характер. Сенильная хорея. Дебютирует в пожилом возрасте, семейный анамнез отсутствует. В клинической картине преобладают гиперкинезы, тогда как когнитивные и эмоциональные нарушения отсутствуют или выражены незначительно. Изолированной формой сенильной хореи является локальный гиперкинез в мимической мускулатуре - орофациальная (или букколингвомастикаторная) дискинезия. Хореический гиперкинез может возникать при целом ряде соматических заболеваний: системной красной волчанке, антифосфолипидном синдроме, сосудистом поражении головного мозга (в области подкорковых ядер), полицитемии, у беременных, принимавших оральные контрацептивы. Прогноз заболевания и особенности терапии определяются, в первую очередь, характером основного патологического процесса. 4.ЛечениеПроводится симптоматическая терапия, направленная на устранение гиперкинезов и купирование психических нарушений. Исходя из сведений о том, что в основе развития хореического гиперкинеза лежит избыточная активность дофаминергической передачи в базальных ганглиях, симптоматическая терапия включает назначение блокаторов дофаминовых рецепторов. Применяют производные фенотиазина, бутирофенона. Одним из наиболее часто назначаемых препаратов является нейролептик галоперидол. Препарат обладает способностью блокировать постсинаптические Д2-дофаминовые рецепторы, кроме того, оказывает блокирующее действие в отношении центральных адренергических рецепторов. Препарат назначается, начиная с 0,5 мг 2 раза в сутки. При необходимости и при хорошей переносимости доза может быть увеличена до 10 мг в сутки. Применение галоперидола сопряжено с риском развития побочных эффектов, вероятность возникновения которых увеличивается с возрастанием дозы препарата. Наиболее частыми побочными эффектами являются двигательные дискинезии (гиперкинезы в мускулатуре лица, конечностей), тремор кистей рук, головы, нижней челюсти, сонливость, заторможенность, задержка мочеиспускания (особенно у пациентов с гиперплазией простаты). Пимозид (производное дифенилбутилпиперидина), который также относится к группе нейролептиков, относительно лучше переносится пациентами, однако, обладает меньшей эффективностью по сравнению с галоперидолом. Основные механизмы действия обусловлены блокадой пре- и постсинаптических дофаминовых рецепторов. Препарат оказывает умеренное гипотензивное, седативное действие. Клинические эффекты применения пимозида наступают достаточно быстро, максимальный эффект отмечается ко второму часу, действие продолжается около 6 часов. Пимозид назначается по 0,5 мг 2 раза в сутки (терапевтическая доза составляет 5,0-8,0 мг в сутки). Препарат противопоказан при сердечных аритмиях, беременности. Применение пимозида ограничивается существенными побочными эффектами, к которым относятся двигательные дискинезии, возможность развития акинетико-ригидного синдрома. Препаратом выбора для лечения больных с болезнью Гентингтона является сульпирид (эглонил) - препарат из группы бензатидов. Относится к группе «атипичных» нейролептиков. Основные механизмы действия сульпирида обусловлены способностью блокировать дофаминовые рецепторы. Характеризуется хорошей переносимостью, редко вызывает расстройства функционирования экстрапирамидной системы. Назначается в суточной дозе 100-300 мг в 2-3 приема; при необходимости доза препарата может быть увеличена. Наиболее частыми побочными эффектами являются нарушения сна, в частности, сонливость или, наоборот, бессонница, оральные гиперкинезы (как проявление дискинезии), нередко – галакторея. Применение препаратов из группы нейролептиков у пациентов с болезнью Гентингтона должно проводиться под постоянным контролем врача, так как существует высокий риск развития акинетико-ригидного синдрома или двигательных дискинезий (гиперкинезов). Вероятность указанных осложнений возрастает при длительном приеме препаратов, а также имеет дозозависимый эффект. Кроме того, при выборе терапевтической тактики следует учитывать вероятность возникновения депрессивных расстройств при применении нейролептиков, что способно привести к значительной социальной дезадаптации пациента. У отдельных пациентов положительный эффект может быть достигнут назначением агонистов центральных постсинаптических α2-адренорецепторов (клонидин, гуанфацин), однако выраженный гипотензивный эффект указанных препаратов в значительной степени ограничивает их широкое применение в клинической практике. У больных ювенильной формой болезни Гентингтона с преобладанием мышечной гипертонии и акинезии показано применение агонистов дофаминовых рецепторов (проноран), дофа-содержащих препаратов (наком, синемет, мадопар), амантадина. Изучается возможность применения препаратов, обладающих нейропротекторным и нейротрофическим действием, для лечения пациентов с болезнью Гентингтона. Эмпирический опыт свидетельствует о наличии положительного эффекта у отдельных больных. Дальнейшие клинические исследования позволят объективно оценить эффективность данного лечебного направления. Попытки хирургического лечения (стереотаксические операции) больных, страдающих хореей Гентингтона, оказались малоэффективными. Разрушение определенных структур базальных ганглиев позволяет у ряда больных добиться временного улучшения состояния в виде купирования гиперкинезов, однако, в последующем наблюдается их возобновление. В последние годы проводятся как экспериментальные, так и клинические исследования возможности применения клеточной терапии пациентов с болезнью Гентингтона. Мультипотентные клетки с заданным вектором выработки нейротрансмиттеров или ростовых факторов вводятся в определенные области головного мозга, где часть из них способна интегрироваться в ткань реципиента и восполнить имеющийся дефицит биологически активных веществ. 5. ПрофилактикаРекомендуется информировать пациентов и ухаживающих за ними лиц/членов семей о целесообразности при необходимости адаптировать места проживания с целью минимизации рисков травматизации пациентов вследствие нарушений контроля произвольных движений (например, убрать ковры, о которые пациент может запнуться, расставить мебель таким образом, чтобы больной мог опираться на неё при передвижении по помещению), а также — на поздних стадиях заболевания, когда развиваются постуральные нарушения, — носить пациентам защитные приспособления при передвижении на длительные расстояния (наколенники, налокотники и пр.) или использовать коляски/ходунков. Рекомендуется уделять достаточно времени при общении с пациентами и членами их семей проведению медикогенетического консультирования. Инвазивная пренатальная генетическая диагностика или преимплантационная генетическая диагностика. Усыновление/удочерение ребёнка. Отказ от деторождения. Применение методов из первой группы даёт возможность носителям мутации БГ иметь родных детей, не являющихся носителями БГ. Таким образом, при БГ МГК позволяет: установить диагноз и тип наследования заболевания в консультируемой семье; определить генетический риск у консультируемых родственников; определить прогноз потомства и наиболее эффективный способ профилактики новых случаев заболевания (в т.ч. с помощью пренатальной и преимплантационной ДНКдиагностики); объяснить консультируемым лицам смысл полученной и проанализированной информации, оказать помощь в решении возникающих юридических, психологических, моральноэтических, социальных и иных проблем. При проведении МГК при БГ рекомендуется следовать следующим подходам. Необходимо предложить пациенту пройти тестирование, представив надлежащую информацию об этом исследовании и самом заболевании, подготовив тем самым обследуемого к возможному положительному результату анализа. Следует предложить пациенту выбрать для себя сопровождающее лицо — человека, который будет сопровождать обследуемого на всех этапах тестирования. Необходимо проинформировать пациента и членов его семьи о том, как положительный результат ДНК-тестирования отразится на существовании потенциального риска носительства мутации другими членами семьи. Следует согласовать план дальнейшего наблюдения за пациентом. Крайне важными при проведении ДНК-тестирования являются получение от обследуемого письменного информированного согласия, подписанного пациентом и врачом, и соблюдение строгой конфиденциальности (так, для участия в процессе медико-генетического консультирования членов его семьи необходимо согласие обследуемого). Пациент имеет право отказаться от сообщения ему результатов ДНК-тестирования или от его проведения вообще. Спустя примерно 1 мес после сообщения положительного результата ДНК-тестирования рекомендуется проведение повторной встречи для обсуждения возможных появившихся у пациента вопросов. В случае положительного результата ДНК-тестирования следует предоставить пациенту более подробную информацию относительно клинических проявлений БГ (или иного подтверждённого заболевания), социальных и психологических последствий выставляемого диагноза, подходов к планированию семьи, доступных на текущий момент методов лечения заболевания. Список использованных источниковГусев, Е.И. Неврология и нейрохирургия/ Е.И. Гусев, А.Н, Коновалов, В.И. Скворцова.-Москва «ГЭОТАР-Медиа», 2015. Гусев, Никифоров, Камчатнов: Неврологические симптомы, синдромы и болезни. Клинические рекомендации. МЗ РФ. МКБ 10: G10.0Возрастная категория: дети, взрослые Год утверждения (частота пересмотра): ID: КР370 2017 https://www.neurology.ru/sites/default/files/assets/documents/2017/07/brochure-hd.pdf?download=1 |