Топохимические реакции. Кинетика топохимических уравнений. Уравнение Ерофеева, уравнение РогинскогоШульца Уравнение сжимающейся сферы
 Скачать 77.64 Kb. Скачать 77.64 Kb.
|

|
Кинетика топохимических уравнений. Уравнение Ерофеева, уравнение Рогинского-Шульца; Уравнение «сжимающейся сферы» СодержаниеКинетические закономерности топохимических реакций 3 Образование зародышей 5 Процессы лимитируемые скоростью образования зародышей 9 Гетерогенные процессы 11 Список литературы 17 Кинетические закономерности топохимических реакцийТопохимические реакции – химические реакции, протекающие на поверхности раздела фаз исходного твердого реагента и образующегося твердого продукта. В них могут участвовать и жидкие, и газообразные фазы. Для таких реакций характерно возникновение границы раздела твердых фаз в результате самой реакции. Типичными топохимическими реакциями являются: разложение солей с образованием твердых оксидов, окисление металлов, дегидратация кристаллогидратов. Основные черты топохимической реакции определяются тем, что реагирующие молекулы, атомы или ионы, образующие кристаллическое вещество, жестко закреплены в кристаллических решетках и лишены той подвижности, которой они обладают в газовой или жидкой фазах. Реакционная способность атомов или ионов в значительной степени зависит от того, в каком месте кристалла они находятся – в объеме, на поверхности грани, на ребре кристалла или на вершине. Кроме того, для топохимической реакции большое значение имеет реальная структура твердого тела, и особенно, наличие дефектов решетки. Очень часто при топохимической реакции продукт реакции сохраняет внешнюю форму кристаллов исходного вещества, вследствие того, что при топохимической реакции возможно лишь минимальное перемещение реагирующих частиц. Топохимическая реакция характеризуется специфическими кинетическими закономерностями. Прежде всего, надо отметить своеобразный характер изменения скорости реакции во времени. В начале реакции ее скорость мала (индукционный период), затем скорость возрастает, достигает максимума и снижается либо до нуля, либо до сравнительно малых значений, и слабо зависит от времени. Интегральная кривая, зависимость степени превращения от времени, представляет собой S-образную (сигмоидную) кривую (рис. 1). Скорость реакции находят как тангенс угла наклона касательной к интегральной кривой при заданном времени(d/d), в результате получают дифференциальную кривую топохимической реакции, зависимость скорости реакции от времени (рис. 2). Используют также понятие эмпирической скорости реакции, которая представляет собой максимальную скорость, соответствующую точке перегиба сигмоидной кривой. Отрезок, отсекаемый на оси времени касательной в точке перегиба интегральной кривой называется периодом индукции. Механизм топохимической реакции включает в себя несколько стадий: образование ядер (зародышей) фазы твердого продукта реакции (период индукции); рост ядер, их перекрывание друг с другом и образование слоя продукта (отрезок после периода индукции до точки перегиба на сигмоидной кривой); увеличение толщины продукта за счет сокращения объема (поверхности) не прореагировавшего вещества (отрезок после точки перегиба). Реакционная поверхность представляет собой поверхность раздела фаз исходного вещества и продукта и называется реакционной зоной или фронтом реакции, ее толщина составляет одну молекулу. Таким образом, ходу кривой скорость-время можно дать следующее объяснение. По мере образования новой фазы твердого продукта в реакции появляется поверхность раздела фаз. Размеры ядер растут, повышается и наблюдаемая скорость реакции. Далее растущие ядра фазы нового продукта начинают сливаться и наблюдаемая скорость реакции проходит через максимум. Продолжающееся слияние отдельных ядер приводит к уменьшению поверхности раздела твердых фаз и скорости реакции. После того, как образуется сплошной слой твердого продукта скорость реакции и по мере уменьшения доли непрреагировавшего вещества реакция прекращается. Образование зародышейДля образования зародыша необходима энергия, которая будет затрачена на перестройку кристаллической решетки и образование межфазовой поверхности. Образование новой фазы (зародыша) внутри исходной материнской фазы относится к фазовым переходам первого рода, которые характеризуются скачкообразным изменением объема, энтальпии и энтропии. На поверхности кристалла S0 имеется z0 потенциальных центров, на которых могут возникнуть ядра нового продукта N. Концентрация таких центров может меняться только за счет образования из них ядер. Тогда скорость образования ядер dN/d можно записать: dN/d = Wуд S0(z0 – N0/S0) (1) Wуд - удельная скорость процесса образования ядер на 1 центр, N0/S0 – уменьшение числа потенциально возможных центров. Проинтегрируем уравнение и разделив переменные получим N = z0S0- exp(-Wуд) (2) Подставив значение N из (2) в (1) и преобразовав, получим dN/d = Wуд exp(-Wуд) (3) Уравнение (3) называют экспоненциальным законом скорости топохимической реакции. Коэффициент Wуд – константой скорости образования зародышей. На ранних стадиях процесса топохимическая реакция хорошо описывается экспоненциальным законом, кроме того, закон справедлив при изотермическом течении процесса и неизменном составе газовой среды. Рост зародышей Для описания стадии роста зародышей и их перекрывания применяют множество уравнений, выбор модели описывающей процесс определяется видом кинетической кривой, выражающей экспериментальные данные, а также формой частиц. Уравнения ускоряющего типа: степенной закон 1/n = k, экспоненциальный закон ln = k. 2. Уравнения сигмоидного типа: уравнения Ерофеева-Авраами: [-ln(1-)]1/2 = k, [-ln(1-)]1/3 = k, [-ln(1-)]1/4 = k. 3. Уравнения кинетики, характеризующиеся диффузионным механизмом: одномерная диффузия 2 = k, двухмерная диффузия (1-)ln(1-) + = k, трехмерная диффузия [1-(1-)1/3]2 = k, уравнение сжимающейся площади 1-(1-)1/2 = k, уравнение сжимающегося объема 1-(1-)1/3 = k, уравнение Гистлинга-Браунштейна [1- 2 /3]-(1-)2/3 = k. Особенность топохимической реакции состоит в том, что она не только локализована на границе раздела фаз, но и в том, что сама граница с течением процесса сама претерпевает изменение, как своей структуры, так и свойств. Топохимическая реакция является сложным процессом. При ее протекании происходит наложение друг на друга вышеперечисленных стадий процесса. При взаимодействии исходного вещества с газом или если продуктом является газ, может возникать внешнедиффузионное или внутридиффузионное торможение. Внешнедиффузионное торможение возникает, когда газообразный исходный реагент или продукт реакции с низкой скоростью подводится или отводится от поверхности реакции. На кривой скорость реакции (d/d) – время образуется площадка. Однако при увеличении скорости газового потока внешнедиффузионное торможение снимается, и кривая 1 преобразуется в кривую 2. Внутридиффузионное торможение возникает в том случае, когда скорость диффузии в порах значительно ниже скорости диффузии газообразных веществ на поверхности вещества, газы поглощаются в устье пор. За счет этого происходит снижение скорости реакции. Чтобы снять внутридиффузионное торможение применяют размельчение частиц, либо используют частицы с более крупными порами, при этом кривая 1 преобразуется в кривую 2. Было обнаружено, что иногда продукты распада при топохимической реакции ускоряют скорость разложения. Это явление проявляется сильнее для измельченных кристаллов, чем для не измельченных. Явление ускорения реакции разложения продуктами реакции называется автокатализом. Фундаментальной характеристикой кинетики твердофазной реакции является энергия активации. Эта оценка широко используется для оценки реакций в газовой и жидкой фазах. Для анализа кинетики реакций в твердой фазе используют экспериментальные данные, пользуясь уравнением k = k0 exp (-E/RT.) Для этого находят зависимость ln k – f(1/T), E = -R [ dlnk/d(1/T)], dlnk/d(1/T) – тангенс угла наклона прямой. Для топохимических реакций энергия активации рассчитывается на некий условный моль. По значению энергии активации судят о том, в какой области протекает процесс. Если энергия активации составляет 5-20 кДж/моль, то процесс лимитируется диффузией, 20-50 кДж/моль переходная область, при энергии активации составляет 50-200 кДж/моль – процесс протекает в кинетической области. Наличие дефектов в структуре материала снижает энергию активации процесса. На скорость реакции влияет размер реагирующих частиц, при измельчении частиц увеличивается поверхность контакта, и следовательно скорость реакции. Введение в состав исходного вещества микродобавок некоторых веществ, называемых легирующими или модифицирующими, также может изменить ход реакции. По правилу Вант-Гоффа, повышение температуры на 10 градусов увеличивает скорость реакции в 2-4 раза. Еще одна возможность активирования реакционных смесей в процессе твердофазного взаимодействия заключается в изменении состава газовой среды. Если реакционные смеси содержат элементы с переменной валентностью, то при изменении окислительного потенциала газовой среды изменяется и состав твердофазных реагентов. Эффективным средством активирования твердофазных процессов является термомеханическая обработка материалов, заключающаяся в одновременном воздействии температуры и давления (прессование). Предполагают, что за счет высокой пластичности материала при повышенных температурах увеличивается площадь контактов между частицами. Например, при получении твердых растворов или ферритов смешивают оксиды или соли нескольких металлов и спекают при повышенных температурах, для улучшения взаимодействия, смесь предварительно прессуют. Процессы лимитируемые скоростью образования зародышейОбразование зародышей новых фаз является обязательной стадией очень многих реакций в твердой фазе и часто может определять их кинетику. Это относится прежде всего к реакциям разложения карбонатов, сульфатов и т. д., изучению которых большое внимание уделяется в СССР, в частности Болдыревым и Павлюченко. Эти реакции являются автокаталитическими и относятся к классу так называемых быстрых реакций в твердых телах, многие из которых, особенно если они сопровождаются выделением энергии, протекают взрывообразно. Для описания кинетики реакций разложения, контролирующийся скоростью образования зародышей, в разное время предлагались различные уравнения. Льюис, затем Центнершвер и Брюсе, Макдональд и Хиншель-вуд безуспешно пытались распространить на эти процессы уравнения кинетики гомогенных, в частности гомогенных автокаталитических, реакций. Рогинский и Шульц, считая, что 1) скорость реакции в твердых телах, идущей через возникновение и рост ядер кристаллического продукта, пропорциональна межфазовой поверхности, на которой локализуется процесс, 2) площадь этой поверхности пропорциональна произведению площадей поверхности исходного вещества и продукта, а также полагая,что S = KV2/3 (где S — площадь поверхности и V — объем фазы), получили для описания кинетики этой реакции выражение 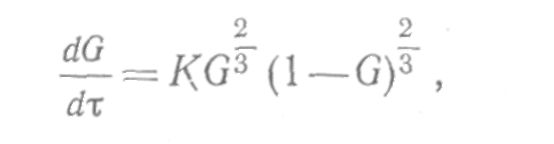 (4) где G — доля прореагировавшего вещества. Справедливость уравнения (4) была подтверждена для процессов разложения КМn04, СаС03-6Н20, оксалата цинка и некоторых других веществ. Тем самым был подтвержден факт локализации подобного рода процессов на поверхности раздела фаз. Впоследствии Ерофеевым было показано, что уравнение (4) не является универсальным и справедливым для всех реакций, идущих через возникновение и рост ядер твердого продукта. Для описания кинетики таких реакций Ерофеев предложил уравнение 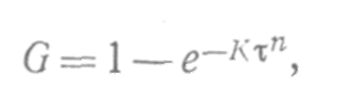 (5) которое можно приблизительно выразить также в виде (6) Эти уравнения выведены в предположении, что процесс возникновения начальных центров реакции может быть многостадийным, т. е. что скорость этого возникновения может зависеть от времени (поскольку концентрация реагирующего вещества не остается постоянной в процессе реакции). Уравнение (5) в развернутом виде выглядит следующим образом: 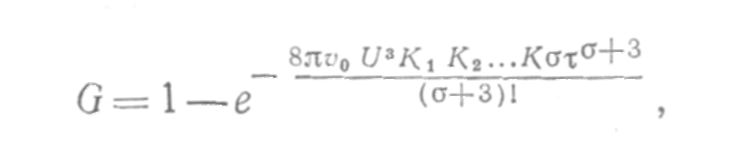 (5а) где v0 — число потенциальных мест, на которых могут возникать начальные центры реакции; о — число промежуточных стадий в процессе образования начальных центров; U — линейная скорость роста ядер реакции; Ki, К2..... Кσ — константы, характеризующие кинетический порядок промежуточных стадий образования начальных центров реакции. Гетерогенные процессыК гетерогенным процессам относятся процессы в системах: газ – твёрдое вещество, газ – жидкость, жидкость – твёрдое вещество, твёрдое – твёрдое, жидкость – жидкость (если жидкости несмешивающиеся), газофазные процессы при участии твёрдых катализаторов. Отличительной особенностью любого гетерогенного процесса является наличие поверхности раздела фаз, которая может быть постоянной, либо меняться во времени. При протекании гетегогенного процесса наряду с чисто химическими стадиями существуют диффузионные стадии. Поэтому для управления гетерогенным процессом важна идентификация лимитирующей стадии. В общем случае для идентификации лимитирующей стадии исследуют зависимость скорости реакции от температуры, и на этой зависимости можно выделить три области: 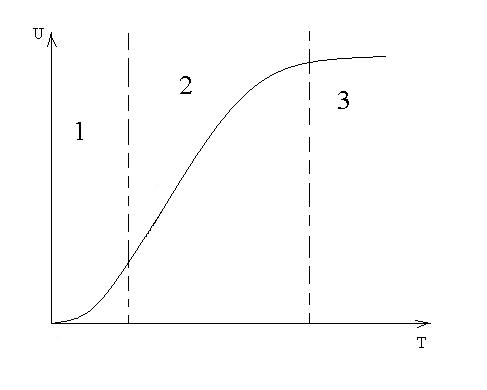 1 – скорость возрастает с увеличением температуры, и выполняется закон Аррениуса. Это кинетическая область протекания процесса. Управляющими будут микрокинетические факторы (температура, давление, концентрации). 3 – скорость процесса практически не зависит от температуры. Диффузионная область энергии активации и диффузии очень мала, следовательно, изменение температуры не приводит к изменению коэффициента диффузии, и скорость изменяется несущественно. Управляющими являются макрокинетическими параметры, связанные со скоростью подачи реагентов, степенью перемешивания реагентов, степенью диспергирования реагентов. Также в этой области в соответствии со скоростью диффузии по первому закону Фика управляющей является концентрация реагентов: 2 – переходная область. Скорость увеличивается с увеличением температуры, но закон Аррениуса не выполняется. В этой области управляющими являются и микро- , и макрокинетические факторы, но интенсивность их воздействия на процесс меньше, чем в соответствующей области протекания. С точки зрения промышленной реализации область 2 наименее перспективна, но следует учитывать, что по ходу гетерогенного процесса он может переходить из одной области в другую. Поэтому для предотвращения перехода изменение одного из микрокинетических параметров обычно сопровождают изменением какого-либо макрокинетического параметра. Гетерогенный процесс – это процесс многостадийный. Наиболее простыми являются процессы в системе жидкость – газ, которые обычно протекают в три стадии: 1-я стадия: область внешней диффузии, то есть подвод газа и жидкости к поверхности раздела фаз, которая чаще всего формируется искусственно (насадочная колонна). 2-я стадия: химическое взаимодействие. Кинетическая область. 3-я стадия: отвод продуктов от поверхности раздела фаз. Внешняя диффузионная область протекания процесса. В подавляющем большинстве случаев процессы в системе жидкость – газ протекают во внешней диффузионной область, поэтому при проектировании оборудования необходимо решать проблему одновременного увеличения линейной скорости подачи реагентов и увеличение площади поверхности насадки. Для того чтобы создать требуемую поверхность контакта фаз необходимо уменьшать размер элементов насадки, что приводит к увеличению гидравлического сопротивления при увеличении скорости подачи реагентов. Следовательно, необходимо искать оптимум в этом вопросе. При проведении адсорбции температура не является управляющим параметром, так как при увеличении температуры растворимость газа в жидкости уменьшается, и увеличение температуры начинают только в том случае, если общая скорость абсорбции лимитируется химической реакцией. Наибольшую сложность для рассмотрения представляют процессы в системе газ – твёрдое вещество. В общем случае процесс можно представить как совокупность 11 стадий: 1-я стадия: диффузия газообразного реагента к поверхности твёрдой частицы (внешняя диффузия). 2-я стадия: диффузия газообразного реагента через слой продукта к поверхности раздела фаз (внутренняя диффузия). 3-я стадия: адсорбция газообразного реагента на поверхности раздела фаз. 4-я стадия: растворение газообразного реагента в твёрдом непрореагировавшем исходном веществе. 5-я стадия: диффузия от поверхности раздела фаз к потенциальному центру образования ядра новой фазы. 6-я стадия: химическая реакция. Далее в обратной последовательности (5-я, 4-я, 3-я, 2-я, 1-я расшифровать). Все 11 стадий наблюдаются в том случае, если уравнение реакции имеет вид: Атв + Вгаз = Ств + Dгаз. Если же: Атв = Ств + Dгаз , то шесть стадий, начиная от химической реакции (с 6-ой по 11-ю). Если: Атв + Вгаз = Ств , то шесть стадий, начиная с диффузии газообразного реагента к поверхности твёрдой частицы (с 1-ой по 6-ю). Для описания кинетики твёрдофазного взаимодействия используется три основные модели образования ядер новой фазы. Первая модель. Образование ядер новой фазы происходит с одинаковой вероятностью на всей внешней поверхности твёрдой частицы при реализации физических условий процесса. Такая модель может быть применена при рассмотрении процессов разложения твёрдого материала, если температура процесса выше, чем температура начала разложения. В этом случае при реализации физических условий процесса вся поверхность твёрдой частицы покрывается слоем продукта, и дальнейшее продвижение к поверхности раздела фаз обуславливается только диффузионными сопротивлениями, обусловленными как пористостью материала, так и размером твёрдых частиц. Вторая модель. Образование ядер новой фазы на активных центрах происходит с одинаковой вероятностью. В качестве активных центров рассматривают дефекты кристаллической решётки твёрдого материала и включения микропримесей, которые обязательно присутствуют в материале. Согласно этой модели считается, что активные центры равномерно распределены по поверхности твёрдой частицы. И при реализации физических условий процесса на поверхности твёрдой частицы образуется фиксированное количество ядер новой фазы. Далее наблюдается рост ядер, что в начальный период времени приводит к увеличению поверхности раздела фаз, а в дальнейшем к её уменьшению. Математически эта модель описывается уравнением сжимающейся сферы: где: x – степень превращения твёрдого материала, k – константа скорости в соответствии с уравнением Аррениуса, Модель сжимающейся сферы наиболее хорошо описывает процессы разложения твёрдого материала и некоторые процессы, связанные с присоединением газообразного реагента. Третья модель. Модель экспоненциального роста числа ядер новой фазы. Универсальна, описывает любой процесс. Предполагает, что активные центры на поверхности твёрдой частицы энергетически неоднородны. При реализации физических условий процесса ядра новой фазы образуются на активных центрах, обладающих наибольшей избыточной энергией. Появление поверхности раздела фаз приводит к активации центров, обладающих меньшей избыточной энергией в первоначальный момент времени. Зависимость скорости процесса от времени обработки твёрдого материала. 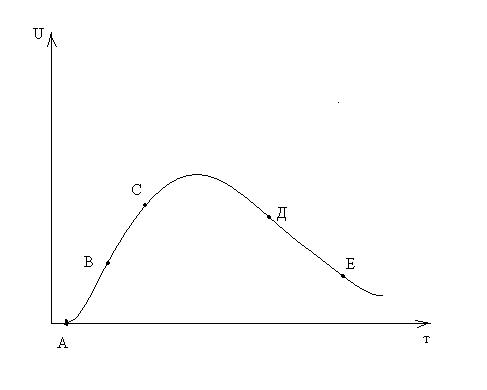 Участок О–А: индукционный период. Он предназначен для накопления энергии в твёрдом веществе. Считается, что в течение индукционного периода протекают первичные превращения, приводящие к возникновению первых ядер новой фазы. Очевидно, что продолжительность индукционного периода зависит от температуры. Участок А–С: период ускорения. На этом участке протекают два параллельных процесса: образование ядер новой фазы и рост уже образовавшихся. Разбит на две части, чтобы показать, что на начальном этапе (участок А–В ) рост связан именно с увеличением количества ядер новой фазы, а на участке В–С – с ростом ядер. Участок С–Д : период максимальной скорости. В точке максимума прекращается образование ядер новой фазы. Участок Д–Е: период спада. В точке Д растущие ядра начинают соприкасаться между собой: из плоских превращаются в шарообразные. С точки Е процесс переходит в диффузионную область. С этой точки вся поверхность частицы покрыта ядрами новой фазы. Эта модель описывается уравнением Ерофеева: n – постоянная Ерофеева. Её физический смысл связан с количеством ядер новой фазы, которое активирует одно образовавшееся ядро. Эта величина определяется экспериментально. Список литературыБайрамов В.М. Основы химической кинетики и катализа / В.М. Байрамов; под ред. В.В.Лунина. М.: Академия, 2003. 256 с Горшков В.И. Основы физической химии / И.А. Кузнецов. М.: БИНОМ, Лаборатория знаний, 2006. 407 с. Ерёмин В.В. Основы физической химии. Теория и задачи: учеб. пособие для вузов / В.В. Еремин [и др.]. М.: Экзамен, 2005. 480 с. Романовский Б.В. Основы химической кинетики: учебник/ Б.В. Романовский. М.: Экзамен, 2006. 415 с. Стромберг А.Г. Физическая химия /А.Г. Стромберг, Д.П. Сем-ченко; под общ. ред. А.Г. Стромберга. 2-е изд., перераб. и доп. М. : Высш. шк., 2001. 496 с. |