Генетика человека с основами медицинской генетики. Классификация наследственных заболеваний и особенности их клинических проявлений
 Скачать 1.3 Mb. Скачать 1.3 Mb.
|

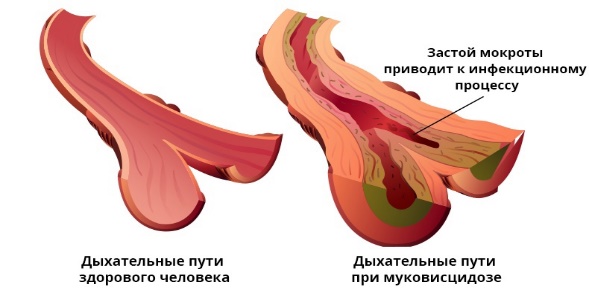
Заболевания с наследственной предрасположенностью или полигенные болезни.К ним относятся: Сахарный диабет; Артериальная гипертензия; Ишемическая болезнь сердца; Ревматоидный полиартрит; Рак молочной железы; Псориаз; Шизофрения; Аллергические заболевания; Язвенная болезнь желудка… Список можно продолжать и дальше. Найдется лишь малая часть болезней, которые так или иначе не связаны с наследственной предрасположенностью. Действительно, все процессы функционирования организма обусловлены синтезом разнообразных белков, как строительных, так и белков-ферментов. 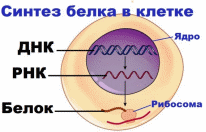 Но если при моногенных наследственных болезней за синтез соответствующего белка отвечает один ген, то при полигенных наследственных заболеваниях за сложный метаболический процесс отвечают несколько разных генов. Поэтому мутация одного из них может быть компенсированной и проявляться только при дополнительных внешних неблагоприятных условиях. Этим объясняется, что у больных данными заболеваниями дети болеют ими не всегда, и, наоборот, у здоровых родителей дети могут болеть этими болезнями. Поэтому в случае полигенных наследственных заболеваний можно говорить лишь о большей или меньшей предрасположенности. 2.1 Генные болезни их особенности Молекулярные болезни - это большая группа заболеваний, в основе которых лежат нарушения в структуре молекул ДНК, т.е. генные мутации. Учитывая последнее, эти заболевания называют также генными болезнями. Молекулярные болезни чрезвычайно разнообразны по характеру клинических симптомов, тяжести течения и прогнозу. Точное количество таких заболеваний не известно, однако, исходя из того, что в организме имеется около 30 тыс. генов и, теоретически, каждый из них может мутировать один или несколько раз, можно допустить, что количество возможных генных бо- 11 лезней будет, если не превышать число генов, то, по крайней мере, соответствовать их сумме. По одной из последних сводок известного генетика Мак-Кьюсика (1988, США) в настоящее время описано 2106 аутосомно-доминантных, 1321 аутосомно-рецессивных и 276 X - сцепленных молекулярных заболеваний. В эту сводку вошли только моногенно наследуемые заболевания, подчиняющиеся менделевским закономерностям. Однако, помимо моногенных заболеваний, имеет место большое число молекулярных болезней с полигенным типом наследования (например, гипертоническая болезнь, атеросклероз, диабет, эпилепсия, шизофрения и многие другие). Для этих заболеваний характерна высокая зависимость не только от особенностей генотипа, но и средовых факторов (стрессы, неправильное питание, инфекции, неизвестные агенты). Эти заболевания получили название мультифакториальных. Для этих заболеваний имеются значительные трудности в определении риска рождения больного ребенка, поскольку наследуемость не подчиняется менделевским правилам. При молекулярных болезнях мутации происходят не только в структурных, но и регуляторных генах. Однако, в любом случае, это сопровождается нарушением синтеза каких-либо белков: ферментных, транспортных или структурных. Если блокируются ферментные белки, развиваются различные патологические состояния, получившие название энзимопатии (греч. "энзим" - фермент, "патос" - страдание, болезнь). Следствием энзимопатии являются болезни обмена. Причем, нарушаться может любой вид обмена: аминокислотный, белковый, углеводный, липидный, минеральный и другие. При болезнях обмена, обусловленных отсутствием какого-либо фермента, снижением его активности или уровня содержания в организме, накапливаются вещества, подлежащие расщеплению (например, продукты промежуточного обмена), или же какие-то аномальные продукты. Накапливаясь в организме, они оказывают на него токсическое действие, что приводит к развитию различных патологических признаков. Клинически выраженные симптомы молекулярных болезней могут проявляться в разные периоды онтогенеза. Некоторые из них диагностируются сразу после рождения, поскольку сопровождаются выраженными внутриутробными пороками развития. Другие - проявляют себя в раннем детстве, третьи - на более поздних периодах индивидуального развития, чаще до наступления репродуктивного (детородного) возраста. Многие генные болезни характеризуются нарушениями не только физического, но и психического развития, и, как правило, в большинстве случаев укорачивают жизнь больного, приводят к частичной или полной инвалидности. Характерный для молекулярных болезней полиморфизм (многообразие) патологических симптомов является следствием не только мутаций, происходящих в разных генах, но и особенностями действия на них геновмодификаторов и различных средовых факторов. В итоге при сходном 12 генотипе патологические признаки имеют различную экспрессивность и пенетрантность даже среди ближайших родственников. В то же время сходные симптомокомплексы могут наблюдаться при мутациях различных генов (генокопии). Это объясняется тем, что разные мутации могут нарушать процесс функционирования одного и того же звена. А мутации в пределах одного и того же гена (множественные аллели) могут приводить к формированию различных фенотипов. Например, метгемоглобинемия - это, в отличие от серповидноклеточной анемии, совершенно другое заболевание, хотя мутация в обоих случаях приходится на один и тот же локус. Общая частота молекулярных болезней в человеческих популяциях составляет 1 -2%. Однако доказано то, что среди детей этот показатель в три раза выше, чем среди взрослых. Условно считают частоту генных болезней высокой, если встречается 1 больной на 10 тыс. новорожденных; средней - в случае, если больной приходится на 10-40 тыс. новорожденных, а далее - низкой. Многие молекулярные болезни могут лечиться при условии, что лечение начато своевременно. В связи с этим разработаны просеивающие программы (скрининг-программы) по раннему выявлению наиболее часто встречающихся генных болезней. Для этого используются простые биохимические методы диагностики, с помощью которых в крови, моче, околоплодных водах при массовых обследованиях определяются аномальные продукты обмена. Скрининговые методы качественные и должны быть специфичными. Важно правильно выбрать время скрининга: в доклинический период развития заболевания, т.е. в досимптомный период, когда возможно эффективное лечение. Кроме чисто биохимических методов, в состав просеивающих программ включают такие специальные методы, как микробиологические тесты, электрофорез, хроматография, радио-иммунологический метод. В практике мирового здравоохранения используются 2 типа просеивающих программ 1.Среди новорожденных на нарушение аминокислотного обмена - аминоацидопатии, галактоземию, гипотиреоз, муковисцидоз и недостаточность α1 – антитрипсина. 2.Среда определенных расовых и национальных групп на гетерозиготное носительство болезни Тея-Сакса, серповидно-клеточной анемии и талассемии. Кроме того, показанием к скринингу у новорожденных в любом случае служат такие нарушения, как судороги, летаргия, кома, трудности кормления, рвота, двигательные беспокойства, необычный запах, катаракта и др. 2.2 Таблица
2.3 Тестовые задания по разделу "Генные заболевания" Наследственные болезни, в основу положен принцип взаимодействия средовых и генетических факторов, относят к: А) морфогенным Б) мультифакториальным В) экзогенным Г) эндогенным Болезни характеризуются тем, что для их развития достаточно унаследовать мутантный аллель от одного родителя А) сцепленные с Х-хромосомой Б) сцепленные с У-хромосомой В) аутосомно-доминантные Г) доминантно-рецессивные Новейшие исследования позволили обнаружить ряд генов, детерминирующий развитие семенников, отвечающий за сперматогенез (фактор азооспермии), контролирующий интенсивность роста тела, конечностей и зубов, оволосение ушной раковины . Эти гены локализованы: А) в У-хромосоме Б) в Х-хромосоме В) в аутосомах Г) правильных ответов нет Аутосомно-доминантная болезнь: организм не в состоянии самостоятельно вырабатывать красные тельца крови. Люди, страдающие этим недугом, не выносят дневного света, т.к. в тканях их организмов нарушен пигментный обмен, и под воздействием солнца происходит распад гемоглобина, который превращается чуть ли не в кислоту и разъедает кожу (кожа больного сильно темнеет и в конечном итоге гниёт и лопается). Превратить "вампира" в нормального человека можно с помощью химиотерапии и частых переливаний крови. В мире насчитывается сегодня около 70 человек, страдающих этим заболеванием. А) прогерия Б) серповидно-клеточная анемия В) синдром Шершевского-Тёрнера Г) порфирия У человека— врожденный порок развития, характеризующийся наличием мужских и женских половых признаков одновременно. А) гермафродитизм Б) вампиризм В) прогерия Г) анахронизм Аутосомно-рецессивное заболевание, как правило, проявляется в детстве, чаще встречается у людей (или их потомков) из тропических и субтропических регионов. Наследственная болезнь человека, при которой красные кровяные клетки (эритроциты), отдавая свой кислород, принимают причудливую удлиненную форму, напоминающую полумесяц или серп. А) фенилкетонурия Б) серповидно-клеточная анемия В) амовратическая идиотия Г) синдром Тея-Сакса Большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена: А) генные болезни Б) геномные болезни В) хромосомные Г) генохромные По типу наследования генные болезни делятся на аутосомно-доминантные, аутосомно-рецессивные, и т.д А) аутосомно-доминантные Б) Х-сцепленные доминантные В) аутосомно-рецессивные Г) все ответы верны В зависимости от системы или органа, наиболее вовлеченного в патологический процесс генные болезни делятся на: А) сердечно-сосудистой системы, мочеполовой системы, желудочно-ки шечного тракта Б) опорно-двигательного аппарата, эндокринные, крови, легких В) нервные, нервно-мышечные, кожные Г) все ответы верны По характеру метаболического дефекта генные болезни делятся на болезни, связанные: А)с нарушением аминокислотного, углеводного, липидного обмена Б) с нарушением минерального обмена В) с нарушением эндокринного аппарата Аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутациями генов, кодирующих синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью. В классических случаях лица имеют удлиненные конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и сердечно-сосудистой системы, А) Синдром Патау Б) Синдром Эдвардса В) Синдром Марфана Г) Синдром Дауна Тип наследования: ХХУ синдром. Клинические признаки: высокий рост, хрупкое телосложение, гипоплазия яичек, импотенция и бесплодие, набухание молочных желез, широкий таз, поперечная ладонная складка, у взрослых наблюдается ожирение и склонность к алкоголизму, незначительное снижение умственного развития. А) синдром Шершевского Б) синдром Клайнфельтера В) синдром "кошачьего крика" Г) синдром Патау
|