Генетика человека с основами медицинской генетики. Классификация наследственных заболеваний и особенности их клинических проявлений
 Скачать 1.3 Mb. Скачать 1.3 Mb.
|

|
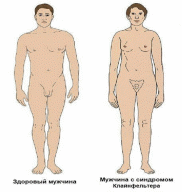
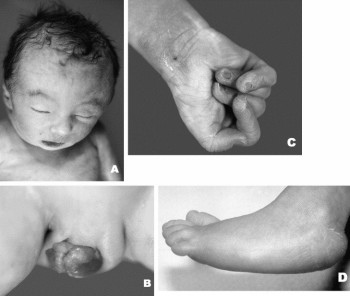
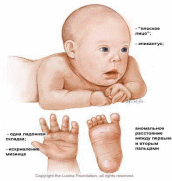
3. Хромосомные заболевания Хромосомные болезни, наследственные заболевания, обусловленные изменением числа или структуры хромосом. Частота Х. б. среди новорождённых детей около 1%. Многие изменения хромосом несовместимы с жизнью и являются частой причиной спонтанных абортов и мертворождений. При спонтанных абортах обнаружено около 20% эмбрионов с аномальными кариотипами (хромосомными наборами). Изменение числа хромосом происходит в результате нерасхождения их в мейозе или при делении клеток на ранней стадии развития оплодотворённого яйца (см. Митоз). Нерасхождению хромосом при первых делениях оплодотворённого яйца способствует, например, высокий возраст матери. Хромосомные аберрации обусловливаются физическими (ионизирующее излучение) и химическими (например, лекарственные препараты с мутагенным эффектом) факторами; вирусами (краснухи, вирусного гепатита, ветряной оспы и др.), антителами и различными расстройствами метаболизма. Х. б. могут быть связаны с излишком генетического материала (полисемия — наличие одной или нескольких добавочных хромосом; полиплоидия; дупликация); с утратой части генетического материала (нуллисомия, моносомия, делеция); с хромосомными перестройками (транслокация; различные перестановки участков хромосом). Различают также группы Х. б., обусловленных изменениями половых и неполовых хромосом. Наиболее распространённые аномалии первой группы у женщин — синдром Шерешевского — Тернера (моносомия Х) и синдром трисомии Х; у мужчин — синдром Клайнфельтера, характеризующийся наличием лишней Х-хромосомы. При синдромах Шерешевского — Тернера и Клайнфельтера возникают задержка полового развития и бесплодие; при синдроме трисомии Х — некоторое снижение интеллекта, расстройства менструального цикла. Частота аномалий по половым хромосомам у мертворождённых составляет 2,7%, что в 25 раз выше, чем среди новорождённых. Среди аутосомных аномалий с нарушением числа хромосом выделяются трисомные синдромы: синдром трисомии хромосом группы D (13—15-е пары), или синдром Патау, встречающийся с частотой 1: 4000 новорождённых; синдром трисомии хромосом группы Е (18-я пара) — Эдвардса, с частотой 1: 300 и Дауна болезнь (трисомия по 21-й хромосоме), частота которой 1: 700 новорождённых. Указанные Х. б. проявляются различными уродствами; задержкой физического и умственного развития; пороками развития внутренних органов. Отмечается специфическое сочетание отдельных аномалий в различных случаях трисомии. Подобные больные живут, как правило, недолго, погибают от вторичных инфекций. Тяжесть клинической картины при синдромах, вызванных структурными изменениями хромосом, как правило, коррелирует с количеством избыточного или недостающего хромосомного материала. Специфика патологических проявлений зависит от того, какая хромосома вовлечена в процесс перестройки. Чаще отмечаются задержка умственного и физического развития, мышечная гипотония, аномалии лицевого скелета. пороки развития внутренних органов. Наряду с типичными Х. б. описано большое количество (около 200) синдромов, вызванных сложными типами хромосомных аберраций. Единственно надёжный метод диагностики Х. б. — цитогенетическое исследование кариотипа, а при изменении числа половых хромосом — дополнительно исследование полового хроматина. Лечение Х. б. сводится к назначению общеукрепляющих, стимулирующих и поддерживающих средств, в том числе гормонов, и др. В профилактике важную роль играет медико-генетическая консультация, которая позволяет выявить семьи с повышенным риском рождения больного ребёнка. Перспективный метод внутриутробной диагностики хромосомного набора плода повышает эффективность медико-генетической консультации в случаях прогнозирования исхода беременности в семьях с повышенным риском рождения ребёнка, больного Х. б. 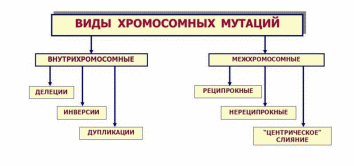 3.1 Перечень хромосомных заболеваний, их особенности и причины Перечень Классификация хромосомных заболеваний основана на нескольких принципах: I. Этиологический, т.е. характеристика хромосомной или геномной мутации: 1) Хромосомные болезни, связанные с аномалиями числа хромосом при сохранении их структуры. – Болезни, обусловленные числовыми аномалиями половых (Х и Y) хромосом (болезни Шерешевского-Тернера, Клайнфельтера). – Болезни, обусловленные числовыми аномалиями аутосом (синдромы Дауна, Патау, Эдвардса). – Болезни, обусловленные увеличением кратности полного гаплоидного набора хромосом – полиплодии. 2) Хромосомные болезни, обусловленные структурными перестройками хромосом. II. Определение типа клеток, в которых возникла мутация (в гаметах или зиготе): – Гаметические мутации ведут к полным формам хромосомных болезней. У таких индивидов все клетки несут унаследованную с гаметой хромосомную аномалию. – Соматические мутации – если аномалия возникает в зиготе или на ранних стадиях дробления, при этом развивается организм с клетками разной хромосомной конституции (два типа и более). Это явление называется мозаицизм, а формы хромосомных болезней – мозаичными. Для того, чтобы мозаичная форма по клинической картине совпадала с полной, необходимо иметь не менее 10 % клеток с аномальным набором. III. Время возникновения мутации (в поколении): – Спорадические случаи – мутация возникла заново в гаметах здоровых родителей или на стадии зиготы. – Наследуемые (семейные) формы – когда родители уже имели подобную аномалию. Таким образом, для точной диагностики хромосомной болезни необходимо определить: 1) тип мутации; 2) вовлеченную в процесс хромосому; 3) форму (полная или мозаичная); 4) вид болезни (спорадический случай или наследуемая форма). Такая диагностика возможна только при цитогенетическом исследовании, проводимом у пациента, а иногда и у его родителей и сибсов. Диагностические признаки хромосомных синдромов можно разделить на три группы: 1. Общие признаки, позволяющие заподозрить аномалии хромосом (психическое или физическое недоразвитие, черепно-лицевой дисморфизм, пороки внутренних органов). 2. Признаки, чаще всего встречающиеся при определенных синдромах. Например, при синдроме Эдвардса в 90 % случаев встречается долихоцефалия и в 96 % – флексорное сгибание кисти. При синдроме Патау в среднем в 70 % случаев встречаются расщелина губы и нёба, микрофтальмия, поликистоз почек, полидактилия. При синдроме Дауна в более 90 % случаев отмечается монголоидный разрез глаз и в 60 % – поперечная складка на ладони. 3. Признаки, патогномоничные для определенного синдрома. Например, при синдроме Лежена отмечается характерный крик, напоминающий кошачье мяуканье, при синдроме де Груши – характерная алопеция. Факторы повышенного риска рождения детей с хромосомными болезнями: 1. Потомство с трисомией появляется у одних и тех же женщин повторно с частотой не менее 1 %. 2. Родственники пробанда с трисомией 21 или другими анеуплоидиями имеют несколько повышенный риск рождения ребенка с анеуплоидией. 3. Кровное родство родителей может повысить риск трисомии у потомства. 4. Резко повышается риск рождения ребенка с трисомией у матери, чей возраст превышает 35 лет. После 45 лет каждая 5 беременность завершается рождением ребенка с хромосомной болезнью. Особенности Этиологическими факторами хромосомной патологии являются все виды хромосомных мутаций и некоторые геномные мутации. У человека встречаются только 3 типа геномных мутаций: тетраплоидия, триплоидия и анеуплоидия. Из всех вариантов анеуплоидий встречаются только трисомии по аутосомам, полисомии по половым хромосомам (три-, тетра- и пентасомии), а из моносомий – только моносомия X. У человека обнаружены все типы хромосомных мутаций: делеции, дупликации, инверсии и транслокации. Делеция (нехватка участка) в одной из гомологичных хромосом означает частичную моносомию по этому участку, а дупликация (удвоение участка) – частичную трисомию. Если транслокация (перенос части хромосомы с одной на другую) является реципрокной (взаимной) без потери участков вовлеченных в нее хромосом, то она называется сбалансированной. Она, как и инверсия (поворот участка хромосомы на 180°), не проявляется у носителя фенотипически, так как при этом сохраняется баланс генов. Однако в процессе кросинговера у носителей сбалансированных транслокаций и инверсий могут образовываться несбалансированные гаметы, то есть гаметы с частичной дисомией, или с частичной нулисомией, или с обеими аномалиями в разных участках. В норме каждая гамета моносомна. При потере двумя акроцентрическими хромосомами коротких плеч и соединении их центромерами может образовываться одна метацентрическая хромосома. Такие транслокации называются робертсоновскими. При концевых делециях обоих плеч хромосомы (делеции теломеров) образуется кольцевая хромосома. У индивида, унаследовавшего такие измененные хромосомы от одного из родителей, будет частичная моносомия по одному или двум концевым участкам хромосомы. Иногда может происходить поперечный, а не продольный, как обычно, разрыв хроматид в области центромер. В этом случае образуются изохромосомы, представляющие собой зеркальное отображение двух одинаковых плеч (длинных или коротких). Наличие у индивида изохромосом проявляется фенотипически, так как имеют место одновременно и частичная моносомия (по отсутствующему плечу), и частичная трисомия (по присутствующему плечу). Хромосомные болезни у новорожденных детей встречаются с частотой примерно 2,4 случая на 1000 родившихся. Большинство хромосомных аномалий (полиплоидии, гаплоидии, трисомии по крупным хромосомам, моносомий) несовместимы с жизнью – эмбрионы и плоды элиминируются из организма матери в основном в ранние сроки беременности. Хромосомные аномалии возникают и в соматических клетках с частотой около 2%. В норме такие клетки элиминируются иммунной системой, если они проявляют себя чужеродно. Однако в некоторых случаях (активация онкогенов) хромосомные аномалии могут быть причиной злокачественного роста. Например, транслокация между 9-й и 22-й хромосомами вызывает миелолейкоз. Патогенез хромосомных болезней еще не ясен. Специфические эффекты связаны с изменением числа структурных генов, кодирующих синтез специфических белков (увеличение при трисомиях и уменьшение при моносомиях). Полуспецифические эффекты при хромосомных болезнях могут быть обусловлены изменением числа генов, представленных и в норме многочисленными копиями. Неспецифические эффекты хромосомных аномалий связывают с содержанием гетерохроматина, играющего важную роль в делении клеток, их росте и других физиологических процессах. Общим для всех форм хромосомных болезней является множественность поражения. Это черепно-лицевые поражения, врожденные пороки развития систем органов, замедленные внутриутробные и постнатальные рост и развитие, отставание в психическом развитии, нарушения функций нервной, иммунной и эндокринной систем. В настоящее время выяснилось, что при хромосомных мутациях наиболее специфичные для того или иного синдрома проявления обусловлены изменениями небольших участков хромосом. Так, специфические симптомы болезни Дауна обнаруживаются при трисомии небольшого сегмента длинного плеча 21-й хромосомы (21q22.1), синдрома кошачьего крика – при делеции средней части короткого плеча 5-й хромосомы (5р15), синдрома Эдвардса – при трисомии сегмента длинного плеча хромосомы. Окончательный диагноз хромосомных болезней устанавливается. Причины Решающим фактором в проявлении хромосомного заболевания является возникновение в гаметах или зиготе на первых этапах её дробления хромосомного нарушения. Схема этих нарушений у человека недостаточно хорошо изучена из-за чрезвычайной сложности изучения влияния внешних и внутренних факторов на гаметогенез и первые дробления оплодотворённой яйцеклетки. К примеру, в мутации в яйцеклетках могут иметь место ещё во внутриутробном периоде развития, поскольку в это время протекает первое мейотическое деление. Фактором, провоцирующим хромосомное нарушение может быть мутагенный фактор физической, химической или биологической природы, действующей в окружающей среде. Иногда мутагенами могут выступать и факторы эндогенного происхождения. Это подтверждают наблюдения за повышенной частотой хромосомных аберраций в организмах при нарушении обмена витамина В12 при некоторых аутоиммунных состояниях. Однако в каждом конкретном случае заболевания выделить мутогенный фактор практически не удаётся и поэтому вернее всего предположить, что такие геномные или хромосомные мутации спонтанны, а не индуцированы. Возникновение хромосомных болезней зависит от возраста, физического здоровья родителей и других факторов. Учёт этих факторов важен для правильного прогнозирования здоровья потомства. Риск иметь ребёнка с трисомией 13, 18 или 21 для женщин в возрасте 40 лет и старше в несколько раз выше, чем у женщин в возрасте 23–25 лет. Механизм такого влияния возраста не выяснен. Влияние возраста матери может быть и обратным: Х-хромосомия чаще встречается у молодых матерей. На примере болезни Дауна обоснована разная роль женского и мужского организмов в рождении детей с трисомией 21: не расхождение хромосомы 21 в мейозе у женщин встречается в 3 раза чаще, а в первом мейотическом делении в 5 раз чаще, чем у мужчин. Если судить по частоте передачи хромосомно несбалансированных гамет от носителей сбалансированных перестроек, между мужчинами и женщинами также имеется существенная разница. Ещё одним внутренним фактором, влияющим на возникновение хромосомного заболевания, является наследственное предрасположение (семейное предрасположение). В семьях, имевших ребёнка хромосомной болезнью при кариотипически нормальных родителях, повторный риск рождения ребёнка с хромосомной патологией хоть и незначителен, но повышен. Известно много подобных случаев, но основные причины остаются до сих пор неясными. Поскольку из экспериментальной цитогенетики известно, что стадии мейоза, включая расхождения хромосом, находятся под генетическим контролем, можно предполагать, что предрасположение к повторному возникновению гамет с численным дисбалансом хромосомного набора также является генетическим.
3.3. Тестовые задания 1. Изучением наследственных заболеваний человека занимается преимущественно: А) медицинская морфология Б) медицинская психология В) медицинская генетика Г) медицинская экология 2. Самостоятельную группу составляют наследственно обусловленные заболевания, возникающие при: А) нарушении функций сердечно-сосудистой системы Б) нарушении минерального обмена В) нарушении обмена веществ Г) несовместимости матери и плода по антигенам групп крови 3. Наиболее типичные черты форм наследственной патологии: •родители обычно здоровы; •чем больше детей в семье, тем чаще встречается более одного больного ребенка; •чем реже встречается мутантный ген в популяции, тем чаще родители больного ребенка являются кровными родственниками; •если больны оба супруга, то все дети будут больными; в браке больного со здоровым рождаются здоровые дети (если здоровый не гетерозиготен); •в браке больного с носителем мутантного аллеля рождается половина больных детей, что имитирует доминантное наследование (псевдодоминирование); •оба пола поражаются одинаково часто. сответствуют наследованию: А) сцепленному с У-хромосомой Б) сцепленному с Х-хромосомой В) аутосомно-доминантному Г) аутосомно-рецессивному 4. При этом типе наследования женщины практически всегда гетерозиготны, т.е. фенотипически нормальны (здоровы) и являются носителями. Больными бывают только мужчины. Речь идет о: А) болезнях с аутосомно-рецессивным типом наследования Б) болезнях с Y-сцепленным доминантным типом наследования В) болезнях с Х-сцепленным рецессивным типом наследования Г) правильных ответов нет 5. Для этого типа наследственности характерны следующие признаки : •болезнь передается только от матери; •больны и девочки, и мальчики; •больные отцы не передают болезни ни дочерям, ни сыновьям. А) митохондриальная наследственность Б) рибосомальная наследственностьсоме В) ядерная наследственность Г) все ответы верны 6. Аутосомно-доминантное заболевание, связанное с появление лишних пальцев на конечностях: А) амовратическая идиотя Б) полидактилия В) арахнодактилия Г) синдактилия 7. Тип наследования: моносомия Х-хромосомы. Клинические признаки: низкий рост, первичная аменорея, бесплодие, стертые вторичные половые признаки, крыловидные кожные складки на шее, врожденные пороки сердца, гипоплазия ногтей, снижение остроты зрения и слуха, поперечная ладонная склад -ка, незначительное снижение умственного развития. А) вампиризм Б) прогерия В) синдром Клайнфельтера Г) синдром Шерршевского-Тёрнера 8. Аутосомно-доминантное наследование. Клинические признаки: недоразвитие или отсутствие одного или нескольких пальцев кистей или стоп. Возможна расщелина губы и неба, умеренная гипоплазия ногтей, неправильная форма зубов, множественный кариес. А) неодактилия Б) полидактилия В) эктродактилия Г) брахидактилия 9. Аутосомно-доминантный тип наследования. Клинические признаки: – это сращение различных пальцев кистей и стоп. На кистях чаще всего встречается между 3 – 4 пальцами, а на стопах - между 2 – 3. А) полидактилия Б) брахидактилия В) синдактилия Г) арахнодактилия 10. Тип наследования аутосомно-доминантный. Клинические признаки: частичная депигментация кожи; поражение обычно симметричное на руках, лице, шее. Больные очень чувствительны к УФ-лучам (получают солнечные ожоги), повышен риск рака кожи. А) синдром Вильямса Б) ахондрогенез В) витилиго Г) гипертрихоз 11. Тип наследования: моносомия по 5-той паре хромосом. Клинические признаки: необычный плач, напоминающий кошачье мяуканье, микроцефалия, антимонголоидный разрез глаз, умственная отсталость, лунопообразное лицо, аномалии внутренних органов. Умирают чаще до 10 летнего возраста. А) порфириоз Б) синдром "кошачьего крика" В) гипертрихоз Г) гипотериоз 12. Одна из форм геномной патологии, при которой чаще всего кариотип представлен 47 хромосомами вместо нормальных 46, поскольку хромосомы 21-й пары, вместо нормальных двух, представлены тремя копиями (трисомия по 21 паре): А) синдром "кошачьего крика" Б) порфирия В) синдром Дауна Г) прогерия
4. Болезни с наследственной предрасположенностью Наследственные болезни – это патологические состояния, в основе которых лежат изменения наследственного материала. Особенности наследственной патологии: 1.Ранняя манифестация: 25% -проявляются сразу после рождения(врожденные); 70% - к 3-му году жизни; 90% - к концу пубертатного периода. 2.Хроническое течение с постоянным ухудшением общего состояния и нарастания негативных симптомов у пациентов 3.Относительная резистентность к терапии 4.Множественность поражения 5.Клинический полиморфизм 6.Семейный характер заболевания Классификация наследственных болезней: 1.Моногенные (генные) болезни – это заболевания, вызванные изменениями в пределах одного гена, т.е. генными мутациями. Они передаются из поколения в поколение и наследуются по законам Менделя. 2.Хромосомные болезни – это заболевания, возникающие в результате хромосомных и геномных мутаций, т.е. связаны с количественными или качественными аномалиями хромосом. 3.Мультифакториальные болезни – это болезни с наследственной предрасположенностью, и для их проявления требуется влияние факторов внешней среды. 4.Генетические соматические болезни – это группа болезней, возникающие в результате мутаций в соматических клетках. К ним относятся: - аутоиммунные заболевания - отдельные пороки развития - некоторые опухоли 5.Болезни генетической несовместимости матери и плода - развиваются в результате иммунологической реакции организма матери на антигены плода. Хромосомные болезни — наследственные заболевания, обусловленные изменением числа или структуры хромосом. Из поколения в поколение передаются не более 3—5 % из них. Хромосомными нарушениями обусловлены примерно 50 % спонтанных абортов и 7 % всех мёртворождений. В настоящее время у человека известно более 700 заболеваний, вызванных изменением числа или структуры хромосом. Около 25 % приходится на аутосомные трисомии, 46 % — на патологию половых хромосом. Структурные перестройки составляют 10,4 %. Среди хромосомных перестроек наиболее часто встречаются транслокации и делеции. Мультифакториальные болезни. Полигенные болезни (ранее — заболевания с наследственной предрасположенностью) обусловлены как наследственными факторами, так и, в значительной степени, факторами внешней среды. Распространение полигенных наследственных заболеваний. Эта группа болезней в настоящее время составляет 92 % от общего числа наследственных патологий человека. С возрастом частота заболеваний возрастает. В детском возрасте процент больных составляет не менее 10 %, а в пожилом — 25-30 %. Распространение мультифакториальных болезней в разных популяциях человека может значительно варьировать, что связано с различием генетических и средовых факторов. В результате генетических процессов, происходящих в человеческих популяциях (отбор, мутации, миграции, дрейф генов), частота генов, определяющих наследственную предрасположенность, может возрастать или уменьшаться вплоть до полной их элиминации. Общие признаки: 1.Неподчинение законам Менделя 2.Высокая частота среди населения 3.Более раннее начало и некоторое усиление клинических проявлений в последующих поколениях. 4.Сходство клинических проявлений болезни у ближайших родственников и пробанда. 5.Клиническое разнообразие. 6.Различная терапевтическая эффективность. 7.Возрастные и половые отличия. Особенности полигенных заболеваний Клиническая картина и тяжесть течения мультифакториальных болезней человека в зависимости от пола и возраста очень различны. Вместе с тем, при всем их разнообразии, выделяют следующие общие особенности: Высокая частота заболеваний в популяции. Так, шизофренией болеют около 1 % населения, сахарным диабетом — 5 %, аллергическими заболеваниями — более 10 %, гипертонией — около 30 %. Клинический полиморфизм заболеваний варьирует от скрытых субклинических форм до ярко выраженных проявлений. Особенности наследования заболеваний не соответствуют менделевским закономерностям. Степень проявления болезни зависит от пола и возраста больного, интенсивности работы его эндокринной системы, неблагоприятных факторов внешней и внутренней среды, например, нерационального питания и др. Кроме того, они связаны с действием многих генов, поэтому их называют также мультифакториальными. Полигенные заболевания тесно связаны с врождёнными дефектами метаболизма, часть из которых может проявляться в виде метаболических заболеваний. Метаболические заболевания — состояние, в котором нормальные метаболические процессы нарушены, чаще всего вследствие отсутствия определённого фермента, или его недостаточности. Более полное определение: группа заболеваний, связанных с нарушениями метаболизма. Причиной заболевания могут быть наследственные нарушения, заболевания эндокринных органов, или иные нарушения в работе метаболически важных органов (например, печени). Генетическое прогнозирование полигенных болезней Генетический прогноз при мультифакториальных заболеваниях зависит от следующих факторов: чем ниже частота болезни в популяции, тем выше риск для родственников пробанда; чем сильнее степень выраженности болезни у пробанда, тем больше риск развития болезни у его родственников; риск для родственников пробанда зависит от степени родства с пораженным членом семьи;, риск для родственников будет выше, если пробанд относится к менее поражаемому полу. подтверждается с помощью генеалогического, близнецового метода-туберкулез, полиомиелит) (ишемическая болезнь сердца, ревматоидный артрит, сахарный диабет, язвенная болезнь, шизофрения 4.1 Особенности болезней с наследственной предрасположенностью (мультифакториальные, полигенные) Семейный характер заболевания. Повторные случаи заболевания в семье указывают на его возможную генетическую природу. Однако единственный случай заболевания в родословной не исключает его наследственной этиологии, поскольку может быть результатом новой (спорадической) мутации или гетерозиготности обоих родителей по рецессивной мутации. Прогредиентное течение – это прогрессирующий характер течения наследственного заболевания. Например, нейродегенеративные заболевания проявляются в виде потери ребёнком ранее приобретенных навыков, приводя впоследствии к развитию тяжёлого поражения ЦНС и гибели больного. Редко встречающиеся специфические симптомы или их сочетание у больного дают основание предполагать наследственную природу заболевания. Например, голубые склеры наблюдаются у больных с несовершенным костеобразованием, грубые черты лица типичны для некоторых болезней накопления, звездчатая радужная оболочка глаз – признак синдрома Вильямса. Врождённый характер заболевания. Если ребёнок рождается с патологическими симптомами, говорят о врождённом характере болезни. Все хромосомные синдромы формируются внутриутробно и могут быть выявлены при рождении, а в некоторых случаях – при ультразвуковом исследовании плода. Тем не менее, врождённость патологических признаков не всегда свидетельствует о наследственной природе заболевания. Примерами врождённых, но не наследственных болезней, являются эмбриофетопатии: талидомидная, алкогольная, вызванная вирусом краснухи и др. Множественное поражение систем органов. Множественное поражение позволяет предполагать наследственную причину заболевания, поскольку большинство мутаций в генах, вызывающих наследственные болезни, даёт плейотропный эффект (см выше). Например, при мутациях гена TSC1, связанных с развитием туберозного склероза, наблюдается ряд признаков – снижение интеллекта, эпилепсия, высыпания на лице в виде, так называемой, аденомы Прингля (представляет собой фиброзную ткань и кровеносные сосуды). Микроаномалии развития у больного. МАР – это необычные морфологические черты, не нарушающие функции каких-либо органов. Они служат индикаторами нарушенного морфогенеза и могут представлять собой симптомы заболеваний. Тщательное описание МАР является одним из диагностических приемов при большинстве наследственных заболеваний. Свыше 40 % известных наследственных синдромов не имеют цитогенетических, биохимических или иных маркеров и идентифицируются на основании фенотипа больного, включая комплекс МАР. Подробно МАР описаны в разделе «Принципы клинической диагностики наследственных болезней» (глава 4.2). Манифестация заболевания в определенном возрасте. Многие наследственные болезни имеют характерный возраст манифестации (от рождения при хромосомных/геномных болезнях до пожилого возраста при болезни Альцгеймера). Так, возраст появления первых признаков фенилкетонурии – 3–4 месяца, а миопатии Эрба-Рота – около 20 лет. Этническая предрасположенность. Некоторые наследственные заболевания встречаются преимущественно у лиц определенных этнических групп. Серповидноклеточная анемия широко распространена в ряде африканских стран, в частности, в Гане. Тирозинемия часто встречается у франкоязычного населения Канады. Устойчивость к терапии. Резистентность к наиболее распространенным методам лечения характерна для наследственных заболеваний. Например, при муковисцидозе респираторная вирусная инфекция приводит к развитию пневмонии, несмотря на лечение. К счастью, резистентность к терапии характерна не для всех наследственных болезней. В тех случаях, когда известен патогенез заболевания, возможна успешная терапевтическая коррекция (например, специфическая диетотерапия при фенилкетонурии и галактоземии, заместительная гормональная терапия при врождённом гипотиреозе). Дополнительными признаками наследственной патологии являются следующие проявления, специфичные для определенных групп заболеваний: спонтанные аборты и мертворождения в семье характерны для хромосомных заболеваний; задержка физического развития типична для более чем 500 моногенных синдромов и для хромосомных аномалий; родственные браки характерны для заболеваний с аутосомно-рецессивным типом наследования; необычный запах мочи и пота – признак ряда метаболических болезней; отставание в психическом развитии является признаком большинства наследственных заболеваний; деменция – ведущий симптом нейродегенеративных заболеваний; нарушение толерантности к физической нагрузке – признак митохондриальных миопатий; увеличение печени и селезенки в сочетании с грубыми чертами лица типичны для болезней накопления. Мультифакториальные заболевания Эта группа болезней отличается от генных болезней тем, что для своего проявления нуждается в действии факторов внешней среды. Среди них также различают моногенные, при которых наследственная предрасположенность обусловлена одним патологически измененным геном, и полигенные. Последние определяются многими генами, которые в нормальном состоянии, но при определенном взаимодействии между собой и с факторами среды создают предрасположение к появлению заболевания. Они называются мулътифакто- риальными заболеваниями (МФЗ). Заболевания моногенные с наследственным предрасположением относительно немногочисленны. К ним применим метод менделев- ского генетического анализа. Учитывая важную роль среды в их проявлении, они рассматриваются как наследственно обусловленные патологические реакции на действие различных внешних факторов (лекарственных препаратов, пищевых добавок, физических и биологических агентов), в основе которых лежит наследственная недостаточность некоторых ферментов. К таким реакциям могут быть отнесены наследственно обусловленная непереносимость сульфаниламидных препаратов, проявляющаяся в гемолизе эритроцитов, повышении температуры при применении общих анестезирующих средств. У человека описана мутация, обусловливающая патологическую реакцию на загрязнение атмосферы, которая проявляется в раннем развитии эмфиземы легких (в возрасте 30—40 лет). У генетически чувствительных индивидов нежелательные реакции могут вызывать некоторые компоненты пищи и пищевые добавки. Известна непереносимость у ряда людей молочного сахара—лактозы. Гены непереносимости лактозы широко распространены среди азиатского населения (до 95—100%) и среди американских негров и индейцев (до 70—75%). У некоторых людей наблюдается непереносимость к употребляемым в пищу конским бобам, вызывающим у них гемолиз. Ряд лиц не переносит жирной пищи и в раннем возрасте страдает атеросклерозом, что повышает риск развития инфаркта миокарда. У некоторых людей употребление в пищу сыра и шоколада провоцирует мигрень. Отмечены специфические реакции людей на алкоголь. Консерванты и пищевые красители у некоторых людей не подвергаются нормальному усвоению, что также проявляется в непереносимости этих компонентов пищи. Наряду с химическими агентами у людей отмечается наследуемая патологическая реакция на физические факторы (тепло, холод, солнечный свет) и факторы биологической природы (вирусные, бактериальные, грибковые инфекции, вакцины). Иногда отмечается наследственная устойчивость к действию биологических агентов. Например гетерозиготы HbA HbS устойчивы к заражению возбудителем тропической малярии. К болезням с наследственной предрасположенностью, обусловленной многими генетическими и средовыми факторами, относятся такие заболевания, как псориаз, сахарный диабет, шизофрения. Этим заболеваниям присущ семейный характер, и участие наследственных факторов в их возникновении не вызывает сомнений. Однако генетическая природа предрасположенности ко многим из них пока не расшифрована. Нередко предрасположенность к ряду заболеваний наблюдается у людей с определенным сочетанием различных генов. Так, у людей со II (А) группой крови чаще наблюдается рак желудка и кишечника, матки, яичников и молочной железы, а также пернициозная анемия, сахарный диабет, ишемическая болезнь сердца, холецистит, желчнокаменная болезнь, ревматизм, а сифилис протекает легче, чем у людей с другими группами крови. У людей с I (0) группой крови чаще встречается язвенная болезнь желудка и двенадцатиперстной кишки. Установление с помощью различных методов генетических исследований точного диагноза заболевания, выяснение роли наследственности и среды в его развитии, определение типа наследования в случае наследственных болезней дают возможность врачу разрабатывать методы лечения и профилактики этих заболеваний в следующих поколениях. Полигенные болезни (ранее — заболевания с наследственной предрасположенностью) обусловлены как наследственными факторами, так и, в значительной степени, факторами внешней среды. Кроме того, они связаны с действием многих генов, поэтому их называют также мультифакториальными. К наиболее часто встречающимся мультифакториальным болезням относятся: ревматоидный артрит, ишемическая болезнь сердца, гипертоническая и язвенная болезни, цирроз печени, сахарный диабет, бронхиальная астма, псориаз, шизофрения и др. Полигенные заболевания тесно связаны с врождёнными дефектами метаболизма, часть из которых может проявляться в виде метаболических заболеваний. |