лекции. Краткий курс лекций структурообразование в пищевых системах воро. Курс лекций структурообразование в пищевых системах
 Скачать 0.85 Mb. Скачать 0.85 Mb.
|

|
Глава 2. УСТОЙЧИВОСТЬ И НАРУШЕНИЕ УСТОЙЧИВОСТИ ДИСПЕРСНЫХ СИСТЕМ Дисперсные системы характеризуются значительным избытком свободной нескомпенсированной поверхностной энергии Gпов = S, связанным как с большой удельной поверхностью S, обусловленной наличием раздробленной дисперсной фазы, так и со сравнительно высокими значениями межфазового поверхностного натяжения. Этот избыток определяет принципиальную неустойчивость лиофобных дисперсных систем. В результате в них протекают процессы, ведущие к снижению дисперсности (укрупнению частиц дисперсной фазы) и, в конечном итоге, к разрушению дисперсных систем, т.е. разделению на две макрофазы. Поэтому речь может идти только об относительной термодинамической устойчивости дисперсных систем. Устойчивость – это способность дисперсных систем сохранять свой состав неизменным, когда концентрация дисперсной фазы и распределение частиц по размерам остаются постоянными во времени. Проблема устойчивости — одна из центральных проблем коллоидной химии, это проблема «жизни и смерти» дисперсных систем. На практике, часто приходится решать две противоположные задачи: сохранить дисперсную систему или разрушить ее. Например, устойчивыми должны быть пищевые массы. Вода природных водоемов представляет собой дисперсную систему, которую для использования необходимо очистить – перевести в осадок все примеси, то есть разрушить. Различают два вида устойчивости: седиментационную и агрегативную. 2.1. Седиментационная устойчивость Седиментационная устойчивость – это способность системы противодействовать оседанию частиц (силе тяжести). Противодействие силе тяжести зависит от размеров частиц. Для крупных (средне- и грубодисперсных) частиц – это сила трения, для мелких (высокодисперсных) – тепловое (броуновское) движение. Повысить седиментационную устойчивость дисперсной системы можно за счет следующих факторов: снижение силы тяжести: достигается путем уменьшения размера частиц с помощью устройств для дробления дисперсной фазы – диспергаторов (уменьшение диаметра частицы в 2 раза приводит к снижению силы тяжести в 8 раз) повышение вязкости среды: достигается при введении различных добавок, повышающих вязкость (сиропы, желатин) обеспечение хранения дисперсной системы при температуре, не ниже установленной нормы, так как при снижении температуры уменьшается броуновское движение, а, следовательно, и седиментационная устойчивость (например, если поставить вино в холодильник, может образоваться осадок). Для нарушения седиментационной устойчивости необходимо наоборот увеличить силу тяжести, для чего применяется центробежное поле (сепарирование, центрифугирование). Например, при сепарировании молока эмульсия разрушается и выделяется молочный жир, капельки которого являются дисперсной фазой системы. 2.2 Агрегативная устойчивость Агрегативная устойчивость – это способность дисперсной системы сохранять неизменной во времени степень дисперсности, то есть размеры частиц и их индивидуальность. При нарушении агрегативной устойчивости происходит слипание частиц и образование крупных агрегатов. В результате система теряет седиментационную устойчивость: частицы не могут принимать участие в броуновском движении и выпадают в осадок. Процесс слипания частиц с образованием крупных агрегатов называют коагуляцией. Укрупнение жидких частиц дисперсной фазы называют коалесценцией. Коагуляция является самопроизвольным процессом, так как она приводит к уменьшению межфазной поверхности и, следовательно, к уменьшению свободной поверхностной энергии. Агрегативная устойчивость и неустойчивость определяются способностью частиц взаимодействовать друг с другом, то есть сближаться до определенного расстояния. Поведение коллоидной системы определяется соотношением сил взаимного притяжения и отталкивания. Между частицами дисперсной фазы действуют силы притяжения (Ван-дер-Ваальса) и силы электростатического отталкивания, обусловленные одинаковым знаком заряда частиц. Энергия притяжения между частицами обратно пропорциональна квадрату расстоянию между частицами h: При сближении двух частиц, несущих одинаковый заряд, происходит перекрытие диффузных слоев противоионов. Возникают силы электростатического отталкивания, энергия которых равна 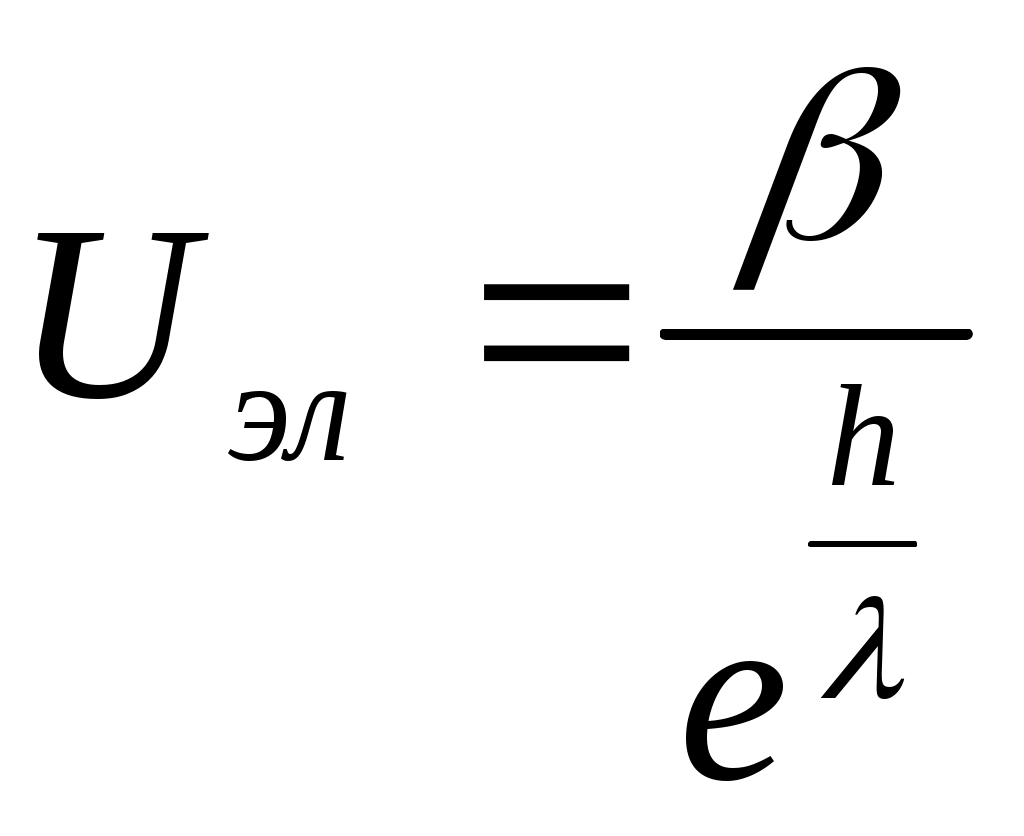 , ,где β – константа, определяемая параметрами ДЭС; h – расстояние между частицами; λ – толщина диффузного слоя. Показатель степени h/λ показывает, во сколько раз расстояние между частицами больше толщины диффузного слоя ДЭС. Суммарная энергия взаимодействия складывается из энергий притяжения и отталкивания: 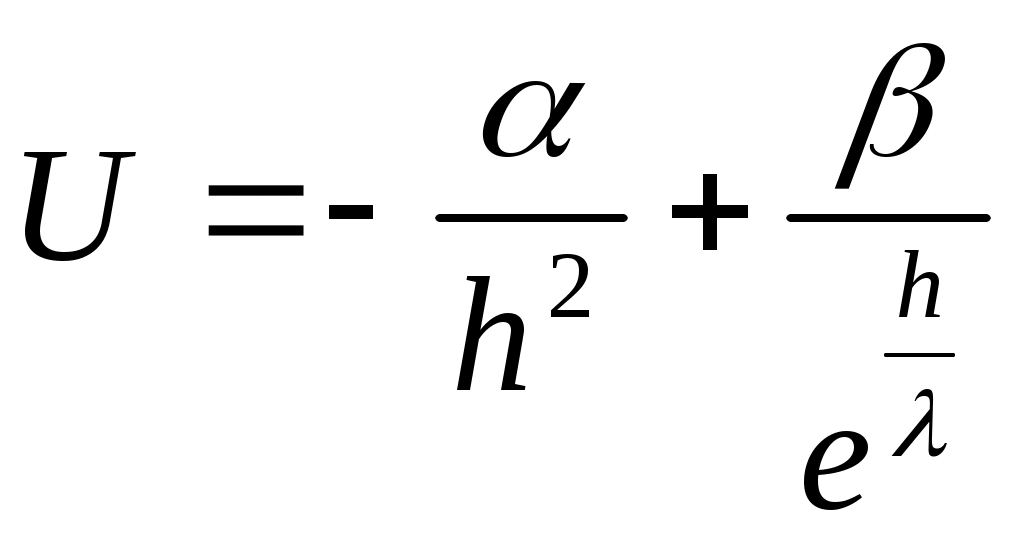 . .Из уравнения следует, что с увеличением h уменьшаются как силы притяжения, так и силы отталкивания. Причем силы отталкивания снижаются значительно резче сил притяжения, так как степенная функция убывает медленнее, чем экспоненциальная. (Например, при увеличении h в 100 раз Uв-в снижается в 104 раз, а Uэл – в 1043 раз.) Зависимость суммарной энергии взаимодействия частиц от расстояния между ними называют потенциальной кривой (рис. 2.1). Так как характер зависимости энергий притяжения и отталкивания от h различный, потенциальная кривая имеет сложный вид. На графике можно выделить три участка: 1 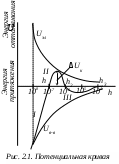 )h < h1; U(h) < 0: между частицами преобладают силы притяжения. При h → 0 энергия притяжения )h < h1; U(h) < 0: между частицами преобладают силы притяжения. При h → 0 энергия притяжения (I min), так называемая первая потенциальная энергетическая яма. Здесь действуют большие силы притяжения, поэтому, если частицы сближаются на расстояния 10-8 – 10-9 м, происходит их слипание и коагуляция. 2) h1 < h < h2; U(h) > 0: основную роль играют силы отталкивания. Возникает потенциальный барьер ΔUк, который не позволяет частицам подходить на близкое расстояние и слипаться. В этом случае коагуляции не происходит, система агрегативно устойчива. 3) h2 < h < h3 U(h) < 0: преобладают силы притяжения. Поскольку силы отталкивания с увеличением h снижаются значительно сильнее, сил притяжения, на относительно больших расстояниях между частицами проявляются дальнодействующие силы притяжения. На потенциальной кривой появляется второй энергетический минимум (II min) или вторая потенциальная энергетическая яма. Величина II min значительно меньше первого. Здесь силы притяжения значительно слабее и взаимодействие частиц происходит через прослойку дисперсионной среды. При этом частицы фиксируются друг относительно друга в пространстве, образуя структурированные системы (неньютоновские жидкости типа гелей и студней). Связи между частицами легко разрушаются, под действием механической нагрузки или температуры, а затем легко восстанавливаются. Факторы, определяющие агрегативную устойчивость коллоидных систем Адсорбционно-ионный фактор. Обусловлен наличием на поверхности частиц двойного электрического слоя и -потенциала. Чем больше силы отталкивания, тем выше потенциальный барьер, и тем более устойчива коллоидная система. Силы отталкивания обусловлены одноименным зарядом частиц, величина которого определяется электрокинетическим потенциалом. Следовательно, чтобы увеличить силы отталкивания необходимо повысить -потенциал. Электрокинетический потенциал тем выше, чем шире ДЭС и выше потенциал ядра φ0. Для того, чтобы увеличить φ0-потенциал, необходимо ввести в золь стабилизатор, т.е. неиндифферентный электролит, повышающий потенциал ядра, а следовательно, и -потенциал. Например, в золь можно добавить небольшое количество NaCl. Так как Сl– и I– изоморфные ионы, то Сl– будет адсорбироваться на поверхности агрегата AgI, в результате φ0 и -потенциал увеличатся. Для повышения -потенциала при неизменном φ0-потенциале необходимо увеличить толщину диффузного слоя , которая пропорциональна: Из уравнения видно, что толщина диффузного слоя увеличивается с повышением температуры и понижением ионной силы раствора. Следовательно, для увеличения устойчивости системы необходимо уменьшить концентрацию противоионов, что достигается при очистке золя. При этом происходит дезагрегация коллоидных частиц (уменьшение размера) или, если выпал осадок, переход его в коллоидное состояние (пептизация). При повышении температуры усиливается броуновское движение частиц, в результате чего устойчивость также увеличивается. Структурно-механический фактор. Заключается в том, что в систему вводят стабилизаторы – высокомолекулярные соединения, ПАВ, которые адсорбируются на поверхности частиц, образуя 3-х-мерную защитную оболочку, не позволяющую частицам сближаться на критическое расстояние. В качестве стабилизаторов в пищевой промышленности применяют желатин и другие белки. Однако введение стабилизаторов не всегда способствует повышению устойчивости. Необходимо правильно подбирать необходимую концентрацию стабилизатора. Если добавить недостаточное количество стабилизатора, то его макромолекула своими концами может адсорбироваться сразу на нескольких частицах, связывая их мостиками (рис. 2.2). В  Рис. 2.2. Образование флокул результате вместо стабилизации происходит укрупнение частиц, и образуются флокулы, которые выпадают в виде рыхлого осадка. Такой процесс называется флокуляцией, а вещества, его вызывающие – флокулянтами. 2.3. Коагуляция золей электролитами Коагуляция – процесс слипания частиц дисперсной фазы с образованием крупных агрегатов. Коагуляция является следствием нарушения агрегативной устойчивости лиофобных золей. В результате коагуляции система теряет свою седиментационную устойчивость, так как частицы становятся слишком крупными и не могут участвовать в броуновском движении. Разрушение коллоидной системы идет в следующей последовательности: Нарушение Нарушение агрегативной → Коагуляция → седиментационной устойчивости устойчивости Коагуляция идет самопроизвольно, так как приводит к уменьшению межфазной поверхности, а следовательно, и к снижению свободной поверхностной энергии (Gпов = S). Различают две стадии коагуляции: медленная (скрытая) коагуляция: частицы укрупняются, но не теряют седиментационной устойчивости; быстрая (видимая) коагуляция: частицы теряют седиментационную устойчивость, при этом, если плотность частиц больше плотности дисперсионной среды, образуется осадок. Количественной характеристикой коагуляции является скорость коагуляции. Скорость коагуляции vк– это изменение концентрации коллоидных частиц Знак “–“ стоит перед производной, так как концентрация частиц со временем уменьшается, а скорость всегда положительна. Чтобы началась коагуляция, частицы должны преодолеть потенциальный барьер ΔUк. Для этого необходимо уменьшить -потенциал. Экспериментально установлено, что коагуляция начинается при 30 мВ. Все сильные электролиты, добавленные к золю в достаточном количестве, вызывают его коагуляцию. При коагуляции золя электролитами различают концентрационную и нейтрализационную коагуляцию. Концентрационная коагуляция Концентрационная коагуляция происходит под действием индифферентного электролита, при этом потеря устойчивости вызывается сжатием диффузной части ДЭС при неизменном потенциале ядра (рис.2.3). 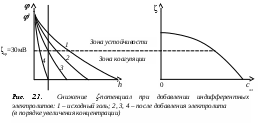 Например, концентрационную коагуляцию положительного золя гидроксида железа будет вызывать NaCl – индифферентный электролит по отношению к данному золю. Скорость коагуляции зависит от концентрации электролита (рис. 2.4.). Зона I – зона устойчивости; при 30 мВ система устойчива, силы электростатического отталкивания преобладают над силами притяжения, коагуляции не наблюдается и скорость ее равна нулю. З 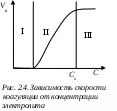 она II – зона медленной коагуляции начинается при достижении -потенциалом критического значения = 30 мВ. Скорость медленной коагуляции зависит от концентрации электролита, поскольку, чем выше концентрация электролита, тем меньше -потенциал, коагуляция идет быстрее. она II – зона медленной коагуляции начинается при достижении -потенциалом критического значения = 30 мВ. Скорость медленной коагуляции зависит от концентрации электролита, поскольку, чем выше концентрация электролита, тем меньше -потенциал, коагуляция идет быстрее. Зона III – зона быстрой коагуляции начинается при приближении -потенциала к 0 (изоэлектрической точке). Скорость быстрой коагуляции не зависит от концентрации электролита. Минимальная концентрация электролита, при которой начинается видимая коагуляция, называют порогом коагуляции Сп. Коагулирующим действием обладает не весь электролит, а только тот ион, заряд которого противоположен заряду коллоидной частицы. Этот ион называется ионом-коагулятором. Коагуляция электролитами подчиняется нескольким эмпирическим правилам. 1. Коагулирующая способность иона-коагулятора тем больше, чем больше заряд иона(первое правило Шульце-Гарди): где α – постоянная для данной системы величина: при z = 1 α = Сп; z – заряд иона-коагулятора. 2. При одинаковом заряде коагулирующая способность иона тем выше, чем больше его кристаллический радиус (второе правило Шульце-Гарди). В соответствии с этим правилом коагулирующая способность иона определяется его положением в лиотропном ряду: одновалентные катионы: Li+ < Na+ < К+ < Rb+ < Cs+ двухвалентные катионы: Mg2+ < Ca2+ < Sr2+ < Ba2+ одновалентные анионы: Сl– < Br– < NQ3– < I– < CNS–. к  оагулирующая способность возрастает оагулирующая способность возрастаетНейтрализационная коагуляция Нейтрализационная коагуляция происходит при добавлении к золю неидифферентного электролита. При этом потенциалобразующие ионы связываются в малорастворимое соединение, что приводит к уменьшению по абсолютной величине термодинамического, а, следовательно, и электрокинетического потенциалов. Когда электрокинетический потенциал достигнет 30 мВ, начнется коагуляция и разрушение дисперсной системы. Рассмотрим в качестве примера положительный золь гидроксида железа: При введении в золь неиндифферентного электролита КОН происходит нейтрализация потенциалопределяющих ионов Fe3+ ионами ОН– с образованием малорастворимого соединения Fe(OH)3. В результате снижается φ0-потенциал и -потенциал, что приводит к ослаблению сил электростатического отталкивания. При снижении до 30 мВ частицы слипаются, и происходит коагуляция. Разновидность нейтрализационной коагуляции – взаимная коагуляция, которая возникает при смешении подобных золей с противоположными знаками зарядов. Например, при смешивании положительно и отрицательно заряженных золей иодида серебра: потенциалопределяющие ионы Ag+ и I– образуют нерастворимое соединение, что приводит к нейтрализации зарядов ядер золей. Потенциал ядра φ0 и -потенциал обоих золей снижаются, происходит коагуляция. |