М. В. Ломоносова Кафедра органической химии Помогаев А. И. Краткий курс органической химии учебное пособие
 Скачать 2.05 Mb. Скачать 2.05 Mb.
|

2. Химические свойства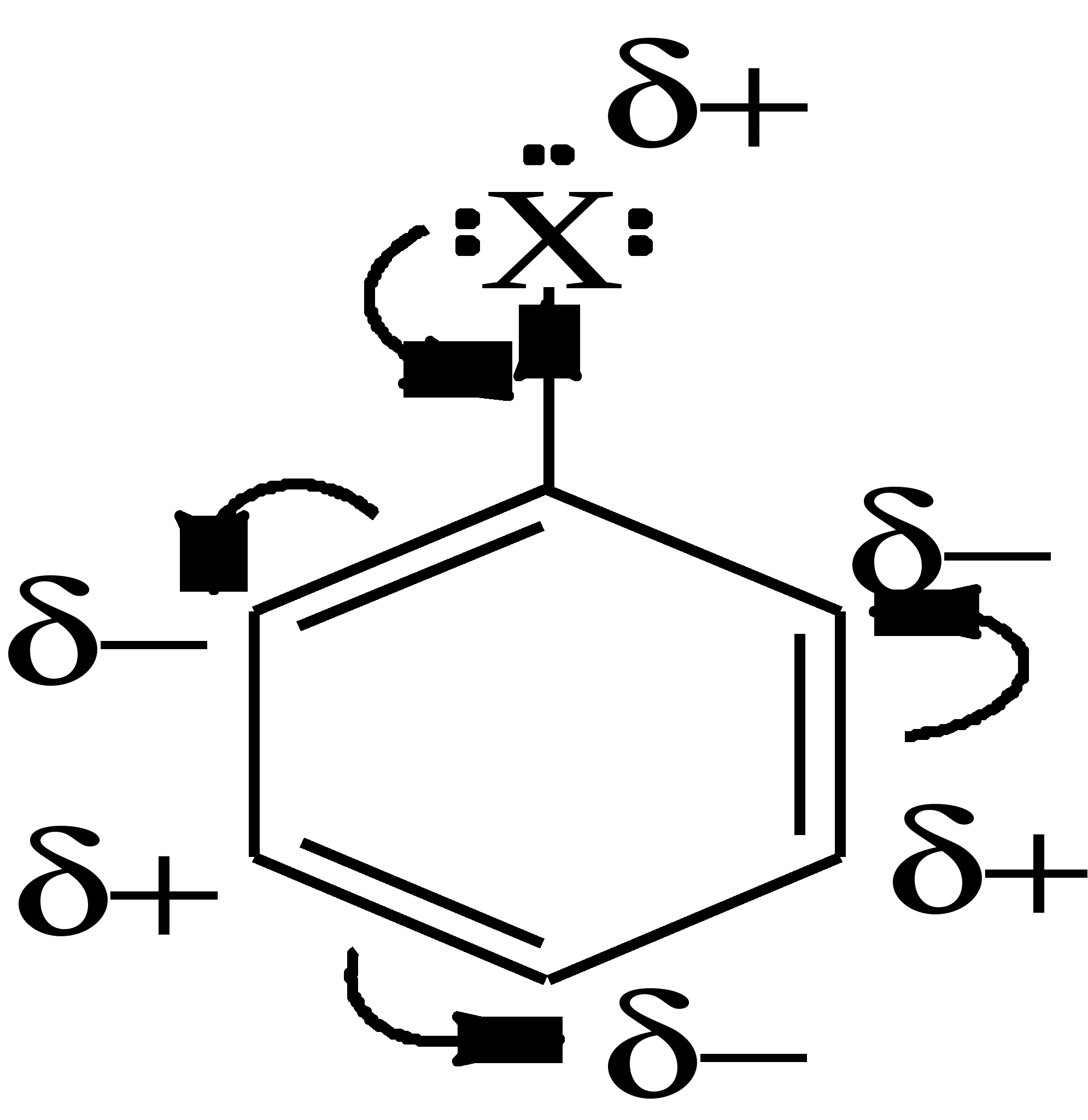 Для галогенопроизводных, имеющих полярную связь углерод-галоген, характерны реакции с гетеролитическим разрывом этой связи, при этом атом галогена уходит как галогенид-анион, т.е. уносит с собой оба электрона связи. В зависимости от строения галогенопроизводного этот разрыв связи может протекать легче или труднее. Для алкилгалогенидов, в которых атом галогена связан с sp3-гибридизированным атомом углерода, этот процесс происходит значительно легче, чем для винил- или арилгалогенидов, у которых атом галогена связан с sp2-гибридизированным атомом углерода не только σ-связью, но и укрепляющим эту связь p-π-взаимодействием. Для галогенопроизводных, имеющих полярную связь углерод-галоген, характерны реакции с гетеролитическим разрывом этой связи, при этом атом галогена уходит как галогенид-анион, т.е. уносит с собой оба электрона связи. В зависимости от строения галогенопроизводного этот разрыв связи может протекать легче или труднее. Для алкилгалогенидов, в которых атом галогена связан с sp3-гибридизированным атомом углерода, этот процесс происходит значительно легче, чем для винил- или арилгалогенидов, у которых атом галогена связан с sp2-гибридизированным атомом углерода не только σ-связью, но и укрепляющим эту связь p-π-взаимодействием.  На место уходящего атома галогена может присоединиться частица, которая образует с остающимся без двух электронов связи атомом углерода новую σ-связь за счет своей пары электронов. Произойдет замещение атома галогена на нуклеофил, т.е. нуклеофильное замещение (SN). Кроме того, уход атома галогена в виде аниона может сопровождаться отрывом протона от соседнего атома углерода, если такой протон имеется. В этом случае происходит отщепление, или элиминирование (Е) галогеноводорода с образованием двойной связи. Обе реакции – и нуклеофильное замещение, и элиминирование – происходят параллельно, т.е. являются конкурирующими, поскольку одна и та же частица может в первом случае выступать как нуклеофил, а во втором случае – как основание, присоединяющее отщепляющийся протон.  нуклеофильное нуклеофильное замещение (SN)  элиминирование (Е) 2.1. Реакции нуклеофильного замещения (SN) в алкилгалогенидахВ реакциях нуклеофильного замещения галогенопроизводные выполняют роль реагента, алкилирующего нуклеофильную частицу. В зависимости от того, несет ли нуклеофильная частица заряд или нет, схемы такого алкилирования могут выглядеть по-разному, например:   В первом случае при взаимодействии этилхлорида с водным раствором едкого натра (щелочном гидролизе этилхлорида) произошло этилирование гидроксид-аниона. Атом кислорода в нуклеофильной частице в процессе реакции выступил донором неподеленной пары электронов и потерял заряд. Во втором случае при взаимодействии трет-бутилбромида с водой (нейтральный гидролиз) произошло алкилирование воды как нуклеофила. Теперь уже нейтральный атом кислорода выступил в качестве донора неподеленной пары электронов и приобрел положительный заряд. Описанный выше гидролиз галогенопроизводных является одним из примеров многочисленных реакций нуклеофильного замещения. В результате гидролиза галогенопроизводных образуются спирты, а в результате реакций с другими нуклеофилами – большое число разнообразных функциональных производных углеводородов. Ниже приведены лишь некоторые другие примеры реакций нуклеофильного замещения:
 бензилхлорид метилат натрия бензилметиловый эфир
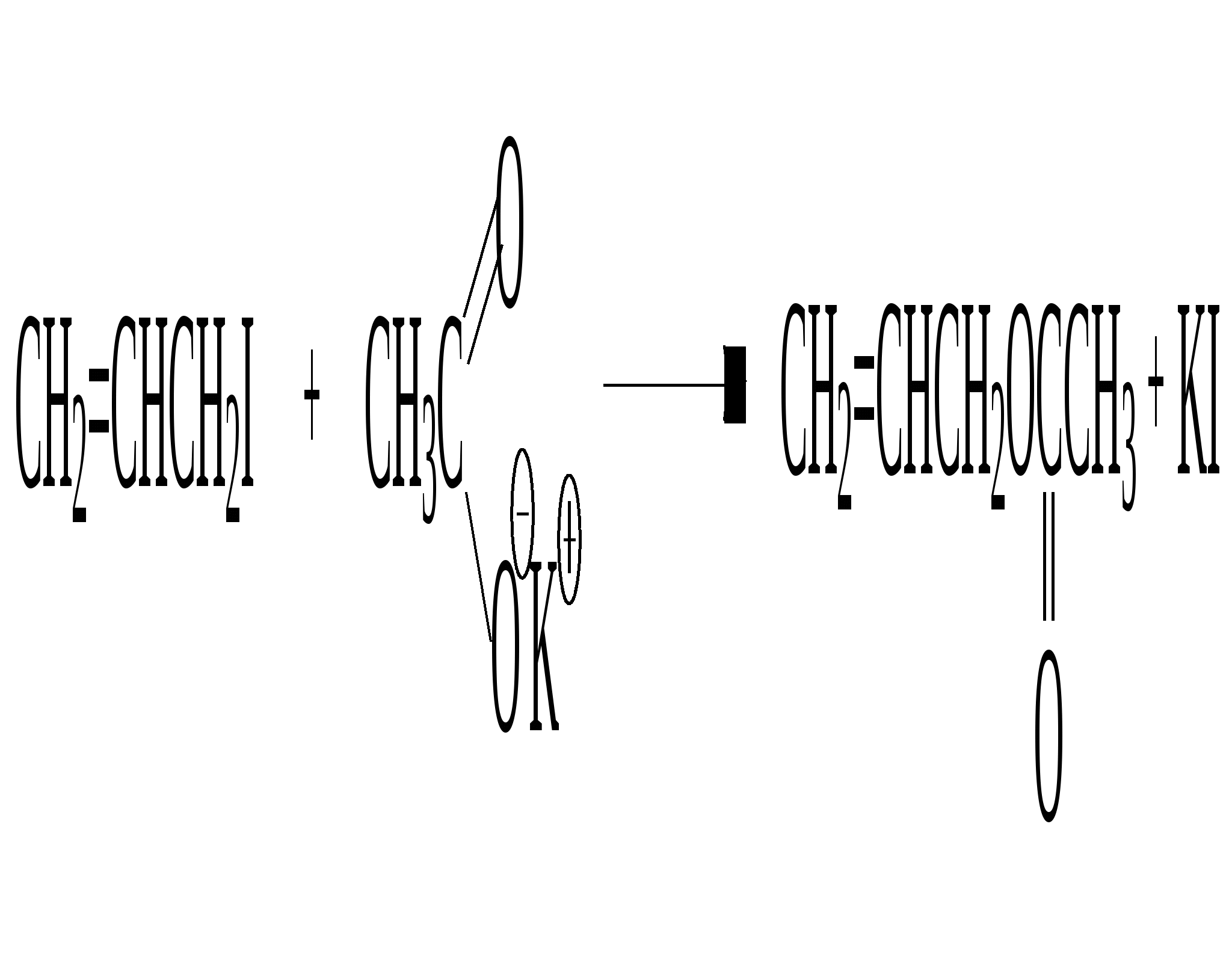 аллилиодид ацетат калия аллилацетат
 иодид метиламмония 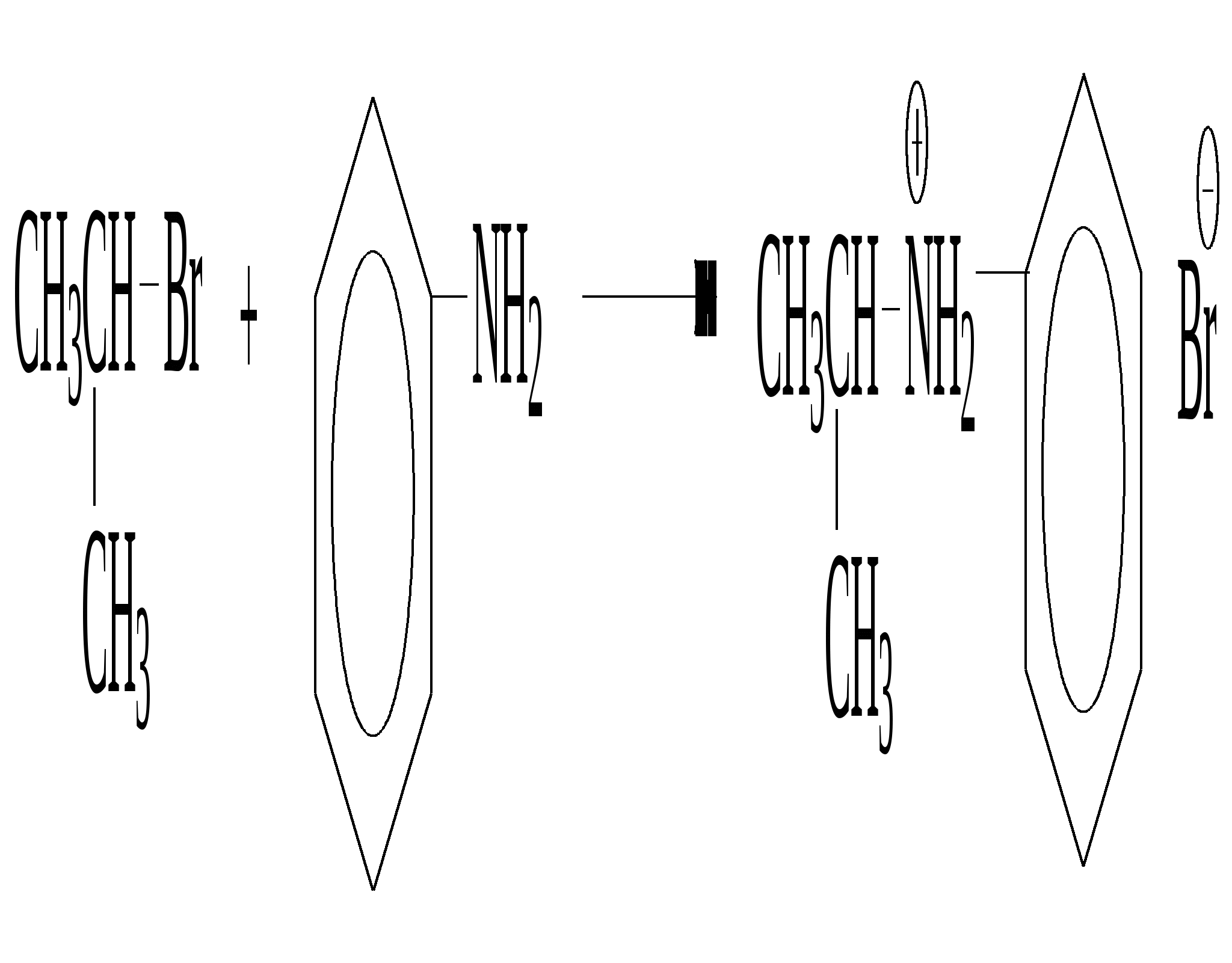 анилин бромид изопропилфениламмония
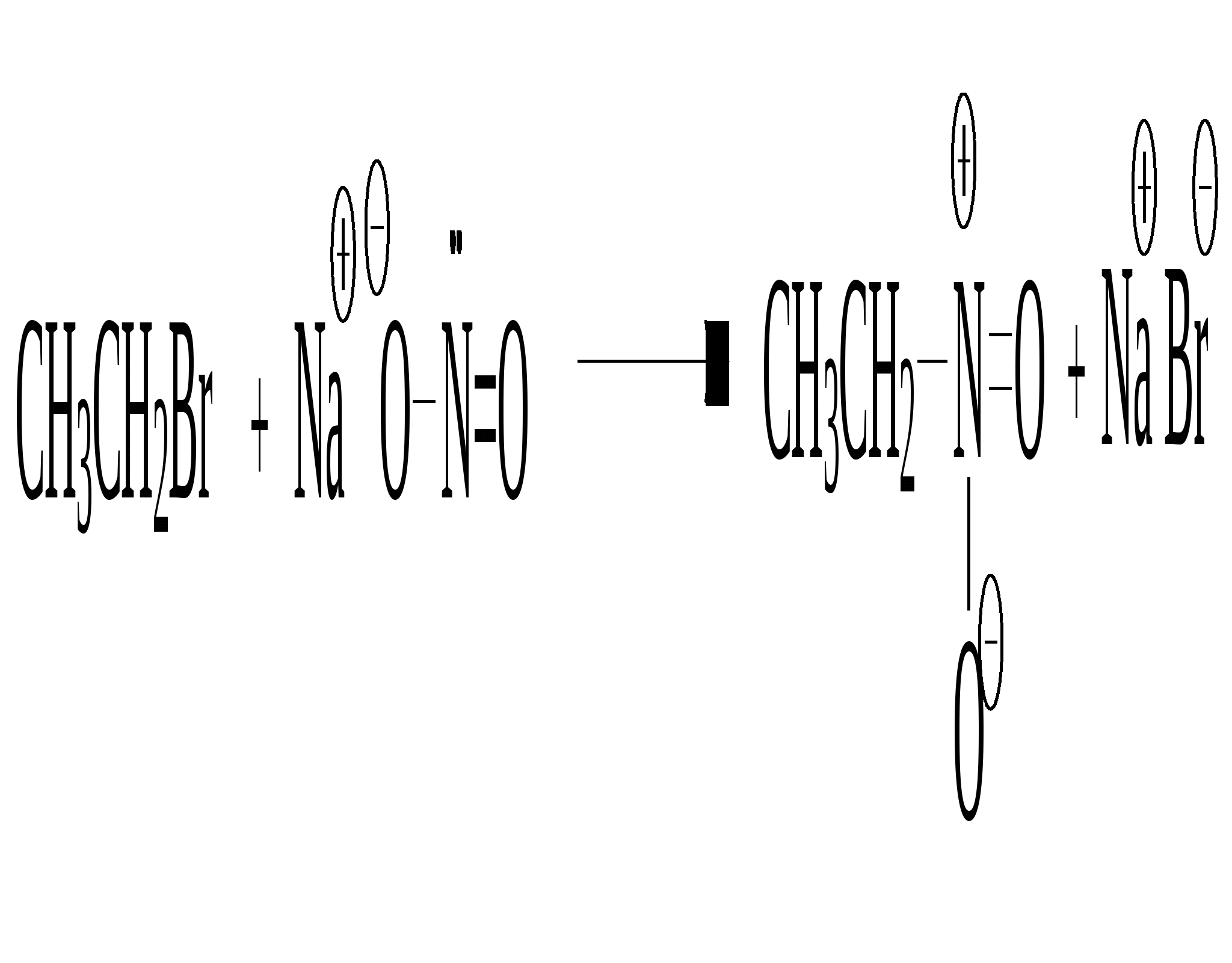 нитроэтан
4-хлор-1-бутен нитрил 4-пентеновой кислоты
метилацетиленид натрия 2-гексин Нуклеофильное замещение в алкилгалогенидах может происходить по двум альтернативным механизмам:
2.1.1. Механизм мономолекулярного нуклеофильного замещения (SN1)Этот механизм представляет собой последовательность двух стадий, отличающихся константами скорости. На первой стадии субстрат медленно подвергается гетеролизу полярной связи углерод-галоген с образованием карбокатиона и галогенид-аниона.  На второй стадии образовавшийся карбокатион очень быстро взаимодействует с нуклеофилом с образованием ковалентной связи по донорно-акцепторному механизму: карбокатион предоставляет вакантную орбиталь, а нуклеофил – заполненную орбиталь.  Так как первая стадия протекает значительно медленнее, чем вторая, скорость реакции замещения определяется скоростью первой стадии, которая зависит только от концентрации галогенопроизводного как единственного участника этой стадии. Такой процесс нуклеофильного замещения является мономолекулярным, поэтому его обозначают SN1. По механизму SN1 реагируют галогенопроизводные, которые при ионизации образуют устойчивые карбокатионы. Поэтому среди насыщенных галогенопроизводных этот механизм характерен, прежде всего, для третичных галогенидов, поскольку третичные карбокатионы являются более устойчивыми, чем вторичные или первичные. Так, например, следующие карбокатионы можно расположить в ряд по убыванию их устойчивости:  Если атом галогена соединен с асимметрическим атомом углерода хиральной молекулы, то мономолекулярное нуклеофильное замещение приводит к рацемизации по данному асимметрическому атому, поскольку образующийся карбокатион как плоская в области заряженного атома углерода частица может присоединить нуклеофил с одинаковой вероятностью с обеих сторон от этой плоскости с образованием двух энантиомеров. Например, при щелочном гидролизе (S)-2,3-диметил-3-хлорпентана образуется смесь равных количеств энантиомерных спиртов, т.е. рацемический спирт.  Кроме третичных галогенопроизводных, по механизму SN1 могут реагировать также аллил- и бензилгалогениды, которые образуют стабилизированные за счет сопряжения карбокатионы. аллил-катион  бензил-катион Поскольку на стадии, лимитирующей скорость реакции, образуются катион и анион, то для протекания реакции по механизму SN1 лучше всего подходят растворители, обладающие возможностью стабилизировать и катионы, и анионы. Такими свойствами обладают так называемые полярные протонные растворители, способные хорошо сольватировать катионы и образовывать водородные связи с анионами. К полярным протонным растворителям относятся, например, вода, муравьиная кислота, а также водно-спиртовые и водно-кислотные смеси. 2.1.2. Механизм бимолекулярного нуклеофильного замещения (SN2)Этот механизм представляет собой процесс нуклеофильного замещения, при котором разрыв связи углерод-галоген и образование связи с нуклеофилом происходят одновременно, а не последовательно, как в случае мономолекулярного замещения. Скорость реакции при этом должна зависеть от концентраций обоих партнеров (и субстрата, и нуклеофильного реагента), поэтому этот синхронный процесс замещения называют бимолекулярным. В момент образования и разрыва связи субстрат и реагент должны пройти так называемое переходное состояние, когда старая связь еще не разорвалась, но уже разрыхляется, т.е. ее электронная плотность уже значительно смещается в сторону атома галогена, а новая связь еще не образовалась, но уже происходит взаимодействие оголяющейся орбитали атома углерода и орбитали с неподеленной парой электронов нуклеофила. Из всех возможных ориентаций субстрата и реагента в переходном состоянии наиболее вероятным является та, которая обладает минимумом энергии. Это реализуется при линейном расположении трех атомов: нуклеофильного центра реагента, атома углерода, при котором происходит замещение, и уходящего атома галогена. В качестве примера приведем механизм взаимодействия метилиодида и аммиака:  переходное состояние Если бимолекулярное нуклеофильное замещение происходит при асимметрическом атоме углерода, то в результате реакции происходит обращение конфигурации. Например, при взаимодействии (S)-2-хлорбутана с ацетатом натрия образуется (R)-втор-бутилацетат.  переходное состояние По механизму SN2 реагируют, прежде всего, галогенопроизводные, не образующие стабильных карбокатионов, т.е. первичные и вторичные. Чем менее разветвлен углеродный скелет вблизи реакционного центра, тем легче замещение протекает по этому механизму. Для протекания реакций по механизму SN2 подходящими растворителями являются диполярные апротонные растворители, такие как ацетон, нитрометан, диметилформамид, диметилсульфоксид и т.п. 2.2. Особенности нуклеофильного замещения галогена в арилгалогенидахСтроение арилгалогенидов, в которых р-π-сопряжение упрочняет связь углерод-галоген, предполагает низкую реакционную способность этих галогенопроизводных в реакциях нуклеофильного замещения. Наличие электроноакцепторных заместителей, которые уменьшают нуклеофильность ароматического ядра, способствует увеличению реакционной способности, т.е. активирует арилгалогениды к реакциям нуклеофильного замещения. 2.2.1. Нуклеофильное замещение в неактивированных арилгалогенидахНеактивированные арилгалогениды реагируют с нуклеофилами в очень жестких условиях. Так, при нагревании до 200 ОС под давлением с избытком разбавленного едкого натра хлорбензол превращается в фенолят натрия. Установлено, что механизм этой реакции включает стадии отщепления хлороводорода с образованием дегидробензола (так называемого бензина) и последующего присоединения к бензину воды. Поскольку фенолы проявляют существенные кислотные свойства (см. ниже), то образующийся при этом фенол реагирует со щелочью с образованием фенолята.  бензин фенол фенолят натрия Подобным же образом происходит взаимодействие хлорбензола и с амидом натрия. При этом в результате нуклеофильного замещения атома хлора на аминогруппу образуется анилин.  анилин 2.2.2. Нуклеофильное замещение в активированных арилгалогенидахЕсли в ароматическом ядре арилгалогенида присутствуют электроноакцепторные заместители, процесс замещения атома галогена на нуклеофил значительно облегчается. При этом реакция протекает по механизму бимолекулярного ароматического нуклеофильного замещения, включающего стадию образования отрицательно заряженного σ-комплекса в результате присоединения нуклеофила и стадию отщепления галогенид-аниона. По такому механизму происходит, например, щелочной гидролиз пара-нитрохлорбензола до пара-нитрофенолята натрия (образующийся при замещении пара-нитрофенол проявляет достаточно сильные кислотные свойства, превращаясь в щелочной среде в пара-нитрофенолят).  σ-комплекс 2.3. Реакция дегидрогалогенированияОтщепление галогеноводорода от молекулы галогенопроизводного происходит региоселективно с образованием наиболее термодинамически устойчивого алкена в соответствии с правилом Зайцева. Так, при нагревании 2-хлорпентана в спиртовом растворе КОН главным продуктом оказывается 2-пентен:  Механизмы, по которым может протекать элиминирование, имеют ту же кинетическую основу, что и механизмы нуклеофильного замещения. Действительно, процесс отщепления заключается в разрыве двух связей в молекуле галогенопроизводного: связи углерод-галоген и связи углерод-водород, причем галоген уходит как анион, а водород – в виде протона, который отрывает основание. Разрывы связей могут происходить либо последовательно, т.е. постадийно, либо синхронно. В первом случае реализуется механизм мономолекулярного элиминирования (Е1), во втором – механизм бимолекулярного элиминирования (Е2). Третичные галогенопроизводные, способные образовать стабильные карбокатионы, подвергаются дегидрогалогенированию по механизму мономолекулярного элиминирования Е1. Так, например, при нагревании трет-бутилхлорида с гидроксидом калия в этаноле происходит сначала ионизация, а затем уже депротонирование образовавшегося на первой стадии карбокатиона:  Для вторичных и первичных галогенопроизводных характерен механизм бимолекулярного элиминирования Е2:  переходное состояние 2.4. Восстановление галогенопроизводныхВосстановление галогенопроизводных до соответствующих углеводородов можно осуществить различными способами:


 2.5. Реакции галогенопроизводных с металламиВ зависимости от природы металлы реагируют с галогенопроизводными по-разному. При воздействии металлического натрия галогенопроизводные по реакции Вюрца превращаются в алканы, например, из этилхлорида образуется бутан. С менее реакционноспособным литием образуются литийорганические соединения, например:  фениллитий При нагревании с магнием в диэтиловом эфире алкил- и арилгалогениды превращаются в магнийорганические соединения – так называемые реактивы Гриньяра:  пропилмагнийиодид  фенилмагнийбромид Металлорганические соединения, такие как литий- и магнийпроизводные углеводородов, являются чрезвычайно реакционноспособными соединениями, которые из-за высокой полярности связи углерод-металл представляют собой сильные С-нуклеофилы.  Реактивы Гриньяра и литийорганические соединения являются также сильными основаниями и реагируют с соединениями, проявляющими кислотные свойства. Поэтому их синтез проводят только в безводных растворителях, поскольку вода, как кислота, реагирует с этими соединениями, гидролизуя их. Подобным же образом реактивы Гриньяра реагируют и с другими более или менее сильными кислотами, например, с карбоновыми кислотами или спиртами:  |