Медицинская генетика. доклад_по_генетике_012007. Медицинская генетика как раздел генетики человека
 Скачать 153.33 Kb. Скачать 153.33 Kb.
|

|
Генетика человека (что изучает генетика человека; методы исследования). Медицинская генетика как раздел генетики человека (рассказать, что изучает медицинская генетика; методы исследования; медико-генетическое консультирование). Моногенные наследственные заболевания человека (рассказать, что это такое; привести несколько примеров заболеваний с кратким описанием; с какой частотой они встречаются в популяции человека). Многофакторные (мультифакториальные) наследственные заболевания человека (рассказать, что это такое; привести несколько примеров заболеваний с кратким описанием; с какой частотой они встречаются в популяции человека). Генетика человека. Мед генетика. - докладчик Лиза Генетика человека наряду с морфологией, физиологией и биохимией является теоретическим фундаментом современной медицины. Она изучает явления наследственности и изменчивости у человека на всех уровнях его организации и существования: молекулярном, клеточном, организменном и популяционном. Генетика человека — раздел генетики, изучающий закономерности наследования и изменчивости признаков у человека. Медицинская генетика изучает роль наследственности в возникновении патологии человека, закономерности передачи от поколения к поколению наследственных болезней, разрабатывает методы диагностики, лечения и профилактики всех форм наследственной патологии. Синтез достижений в медицине и генетике направлен на борьбу с болезнями и улучшение здоровья людей. Медицинская генетика отвечает на следующие конкретные вопросы: какие наследственные механизмы поддерживают гомеос-таз организма и определяют здоровье индивида; каково значение наследственных факторов в этиологии болезней; каково соотношение наследственных и средовых (ненаследственных) факторов в патогенезе болезней; какова роль наследственных факторов в определении клинической картины болезней (и наследственных, и ненаследственных); влияет ли (и если влияет, то как) наследственная конституция на процесс выздоровления и на исход болезни; какие наследственные факторы определяют специфику фармакологического и других видов лечения. В настоящее время медицинская генетика интенсивно развивается в разных направлениях: изучение генома человека, цитогенетика, молекулярная и биохимическая генетика, иммуногенетика, генетика развития, популяционная генетика, клиническая генетика, экологическая генетика. Значение генетики для медицины трудно переоценить. Во-первых, как часть теоретического фундамента медицины, генетика расширяет и углубляет биологическое мышление специалиста. Будущий медицинский работник через понимание законов наследственности и изменчивости реально представляет все стадии индивидуального развития человека (от оплодотворения до старости) под углом зрения реализации унаследованной индивидом программы в конкретных условиях среды. Генетические знания необходимы для понимания новых методов диагностики, лечения и профилактики наследственных болезней, создания новых вакцин и лекарств методами генетической инженерии. клинической дисциплины, эффективно внедряются во все разделы медицинской помощи и здравоохранения (больничная, поликлиническая, диспансерная службы). Наследственные болезни занимают существенное место в работе каждого врача и медицинской сестры в связи с их частотой и тяжестью. Известно уже около 20 000 наследственных признаков, почти 5000 из которых составляют наследственные болезни, поражающие все органы, системы и функции организма. Около 5% детей рождается с наследственными и врожденными болезнями. ЗАДАЧИ МЕДИЦИНСКОЙ ГЕНЕТИКИ (со слайда) Методы медицинской генетики (со слайда) Клинико-генеалогический метод был предложен в 1865 г. Гальтоном, однако как метод изучения наследственности человека его стали применять только с начала XX столетия. Клинико-генеалогический метод дает возможность: • выявлять наследственный характер признака; • определять тип наследования; • определять пенетрантность гена; • изучать закономерности мутирования отдельных генов; 7 • устанавливать носительство мутантного гена тем или иным членом семьи; • определять вероятность генетически обусловленных событий и рассчитывать риск наследования патологического гена (признака) при медико-генетическом консультировании. Клинико-генеалогический метод лежит в основе медико-генетического консультирования и включает 3 этапа: 1 этап – сбор генетической информации;(Проводится анкетирование,опрос. Чем больше поколений удается охватить, тем вероятнее получение достоверной информации) В родословную вносят сведения о выкидышах, абортах, мертворожденных, бесплодных браках, внебрачных детях и др. При сборе информации необходимо внимательно анализировать сообщения об инфекциях и травмах 2 этап – составление родословной; (На основании полученных данных составляется графическое изображение родословной.) На рисунке 1 изображены стандартные символы, применяемые при составлении родословных (предложено в 1931 г. Г. Юстом). (При составлении графического изображения родословной важно соблюдать следующие правила: 1. Составление родословной начинают с пробанда. Братья и сестры (сибсы) располагаются в порядке рождения слева направо, начиная со старшего.) 2. Все члены родословной располагаются строго по поколениям, в один ряд. 3. Поколения обозначаются римскими цифрами слева от родословной сверху вниз. 4. Арабскими цифрами нумеруется потомство одного поколения (одного ряда) слева направо. Благодаря такой нумерации каждый член семьи имеет свой шифр (например: I-1, I-2, II-2, II-4 и др.) 5. Указывается возраст членов семьи (родословной), в связи с тем, что некоторые болезни проявляются в разные периоды жизни. 6. Отмечаются лично обследованные члены родословной. 3 этап – генетический анализ родословной Задача генетического анализа – установление наследственного характера заболевания и типа наследования, выявление гетерозиготных носителей мутационного гена, установление генотипа пробанда. Первая задача при анализе родословной – установление наследственного характера признака. Если в родословной встречается один и тот же признак (или болезнь) несколько раз, то можно сделать предположение о его наследственной природе. После того как будет обнаружен наследственный характер признака (болезни), необходимо установить тип наследования. Кроме того, анализ родословной позволяет установить предполагаемое носительство мутантного гена членами родословной, а также генетический риск рождения больного ребенка у разных членов семьи. Медико-генетическое консультирование – специализированный вид медицинской помощи населению направленный на профилактику наследственных болезней. Задачи МГК • установление точного диагноза наследственного заболевания, которое явилось поводом обращения к генетику; • определение типа наследования заболевания, • выбор наиболее эффективного способа его профилактики у будущего ребенка • объяснение родителям значения собранной информации для постановки диагноза, медикогенетического прогноза и методов профилактики Перечь медицинских показаний для проведения медико-генетического консультирования и диагностики Моногенные заболевания - докладчик Лаура Моногенные болезни — разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями на уровне гена. Закономерности наследования моногенных болезней соответствуют законам Менделя. В настоящее время описано более 4000 моногенных болезней. Этиологическим фактором генных болезней является мутация на уровне ДНК. Мутации, вызывающие наследственные болезни, могут затрагивать ферменты и структурные, транспортные и рибосомальные белки. В каждом гене может возникать до нескольких сотен вариантов мутаций (разные типы в разных участках гена). Любая из вышеперечисленных мутаций может привести к наследственным болезням. Мутации могут реализовываться в различные периоды онтогенеза: до 25 % наследственной патологии проявляется внутриутробно, в до пубертатном периоде — 45 %, в подростковом и юношеском периоде — 20%, и лишь 10% моногенных болезней развиваются в возрасте старше 20 лет. Моногенные заболевания, их патогенез, диагностика и лечение Адреногенитальный синдром (врожденная гиперплазия коры надпочечников). Выделяют 5 типов синдрома, различающихся биохимическим дефектом стероидогенеза. Частота заболевания 1:5000 новорожденных. Наиболее распространенный тип заболевания обусловлен недостаточностью 21-гидроксилазы, участвующей в синтезе кортикостероидов. Тип наследования: аутосомно-рецессивный. Недостаточность 21-гидроксилазы приводит к снижению содержания в крови альдостерона и кортизола. Снижение концентрации кортизола но принципу обратной связи стимулирует выработку адренокортикотропного гормона. Высокий уровень гормона усиливает выработку андрогенов и приводит к гиперплазии тех зон коры надпочечников, в которых синтез гормонов не нарушен. В зависимости от степени недостаточности 21 -гидроксилазы различают две клинические формы заболевания. При вирилизирующей форме избыточная продукция андрогенов во внутриутробном периоде у новорожденных девочек приводит к различной степени маскулинизации от умеренной гипертрофии клитора до полного срастания губно-мошоночных складок с фор- жированием предстательной железы, мошонки, пенисообразного клитора. У мальчиков отмечается раннее половое развитие и низкий рост. Отмечается гиперпигментация гениталий, кожи вокруг сосков, кожных складок. Костный возраст опережает паспортный. Характерен низкий рост, обусловленный ранним закрытием зон роста. При сольтеряющей форме, обусловленной полным дефицитом фермента, на первый план выступают рвота, тахикардия, признаки дегидратации, гипонатриемия, гиперкалиемия. При гормональном исследовании выявляют: высокую концентрацию 17-гидроксипрогестерона, тестостерона, адренокортикотропного гормона, снижение концентрации кортизола, альдостерона в крови, увеличение содержания 17-кетостероидов в моче. Диагноз основан на результатах гормонального обследования и данных молекулярно-генетического исследования. Дифференциальный диагноз проводится с другими формами недостаточности коры надпочечников. Лечение проводится посредством гормонозаместительной терапии. Профилактика возможна путем массового скрининга новорожденных, выявления гетерозигот в пораженных семьях с помощью нагрузочных тестов, пренатальной диагностики. Муковисцидоз (кистофиброз). Частота заболевания колеблется от 1:1700 до 1:3000 новорожденных. Причиной возникновения заболевания являются мутации в гене транспортного белка хлоридного канала в эпителиальных клетках экзокринных желез (респираторного и желудочно-кишечного трактов, потовых, поджелудочной желез и др.) (ген картирован на 7q31). Тип наследования: аутосомно-рецессивный. Недостаточность транспортного белка хлоридного канала приводит к нарушению электролитного обмена в клетках легких, кишечника, потовых, поджелудочной, слюнных железах, изменению состава и свойств секретируемых жидкостей. Существует несколько клинических форм заболевания: кишечная форма, легочная форма, легочно-кишечная форма и мекониальный илеус у новорожденных. Мекониальный илеус (неспособность продвижения мекония по кишечнику) встречается у 5-10% новорожденных с муковисцидозом. Основные клинические проявления: рвота желчью, большой выступающий живот, боль в животе, перфорация кишечника. В тяжелых случаях — перитонит, приводящий к летальному исходу. Легочная форма заболевания проявляется на первом году жизни. Отмечаются кашель, сопровождающийся отделением вязкой гнойной мокроты, дыхательная недостаточность, стридор, бронхиты с выраженным бронхоспазмом, пневмонии. Типичным признаком заболевания является пневмосклероз с бронхо- эктазами. С возрастом заболевание неуклонно прогрессирует. При кишечной форме на первый план выступают признаки кишечной мальабсорбции в форме стеатореи и гнилостного характера стула. Для диагностики заболевания проводят определение концентрации хлоридов пота (50-180 ммоль/л, при норме менее 40 ммоль/л) и молекулярно-генетическое исследование. Дифференциальный диагноз проводят с целиакией, другими бронхолегочными заболеваниями. Лечение симптоматическое: антибиотико-, ферменто-, витамино-, кинези-, диетотерапия, муколитическая и бронхолитическая терапия. Профилактика возможна путем скрининга новорожденных на муковисцидоз, пренатальной диагностики с помощью молекулярно-генетического анализа. Несовершенный остеогенез. Несовершенный остеогенез — это гетерогенная группа наследственных заболеваний соединительной ткани, характеризующиеся повышенной ломкостью костей. Суммарная частота составляет 1:20 000. Типы наследования: аутосомно-доминантный (I и IV типы) и аутосомно-рецессивный (II и III типы). Заболевание обусловлено мутациями структурных генов COL1A1 и COL1A2 а-цепей коллагенов (гены картированы для а, (I) цени — на 17q21-q22, для а2 (I) картирован на 7q21-q22). Мутации в генах а-цепей коллагена первого типа приводят к нарушению его свойств и структуры тканей, содержащих этот коллаген. Основными клиническими признаками заболевания являются переломы длинных трубчатых костей, реже ребер и ключиц, голубые склеры, снижение слуха. Описаны переломы фаланг пальцев и других костей. Для несовершенного остеогенеза характерны опалесцирующие («янтарные») зубы, гипермобильность суставов, пролапс митрального клапана, деформация грудной клетки, кифосколиоз, остеопороз, грыжи. Наличие у больного переломов костей, возникших внутриутробно, а также переломов костей при рождении, особенно в сочетании с голубыми склерами, позволяет заподозрить несовершенный остеогенез. Снижение слуха редко развивается ранее 10-летнего возраста. Пенетрантность заболевания неполная. Пораженные индивиды могут иметь только один из основных клинических признаков заболевания. В данном случае только тщательно проведенный клинико генеалогический анализ помогает поставить правильный диагноз. Диагноз основан на совокупности клинической картины и клинико-генеалогического анализа. Дифференциальный диагноз проводится с другими скелетными дисплазиями, сопровождающимися деминерализацией костей. Ахондроплазия. Ахондроплазия — наследственное заболевание костной системы, характеризующееся ризомелической формой карликовости. Частота заболевания 1:100 000 новорожденных. Тип наследования: аутосомно-доминантный. Более чем 70 % случаев представлено свежими мутациями. Заболевание обусловлено точечными мутациями в гене коллагена COL2A1 (ген картирован на 12ql3-ql4). Ведущими клиническими признаками заболевания являются диспропорциональная карликовость, укорочение проксимальных отделов конечностей, макроцефалия, запавшая переносица, выраженный поясничный лордоз, изменения костей таза, изменения позвоночника (сужение расстояния между корнями дужек поясничных позвонков, нарастающих в каудальном направлении). Характерны широкие кисти, пальцы в виде трезубца, изодактилия. Средний рост при рождении составляет 47 см, а средний рост для взрослых составляет 130 см для мужчин и 123 см для женщин. Дети, как правило, отстают в моторном развитии, интеллект нормальный. Встречаются гидроцефалия, описаны частые отиты. Диагноз ставится на основании характерной клинической картины. Дифференциальный диагноз проводится с различными типами ахондрогенеза. Синдром Холта—Орама (синдром руки-сердца) Основными клиническими симптомами являются ВПР верхних конечностей (чаще поражается левая рука): гипоплазия, отсутствие, удвоение, дигитализация I пальца кисти, трехфаланговый I палец. Характерны изменения других пальцев (клинодактилии, синдактилии), гипоплазии лучевой, локтевой, плечевой костей. Описаны аномалии ключиц, лопаток, различные деформации грудной клетки, кифоз, сколиоз. В 85% случаев выявляют пороки сердца: дефекты межпредсердной и межжелудочковой перегородок, открытый артериальный проток, коарктация аорты. В остальных случаях при отсутствии структурного дефекта выявляют аномалии ЭКГ. Диагноз ставится на основании специфической клинической картины. Дифференциальный диагноз проводится с синдромом панцитопении Фанкони, тромбоцитопении с отсутствием лучевой кости, дефектами лучевой кости, VACTERL-ассоциацией, другими типами синдромов рука-сердце. Наиболее часто встречающимися моногенными заболеваниями из группы факоматозов являются нейрофиброматоз I типа (болезнь Реклингхаузена) и туберозный склероз. Клиническими симптомами, характерными для этой группы заболеваний, являются пигментные пятна и склонность к образованию опухолей. Нейрофиброматоз I типа (болезнь Реклингхаузена) Нейрофиброматоз I типа является самым частым наследственным заболеванием из группы факоматозов, характеризующимся предрасположенностью к возникновению опухолей периневрия. Встречается с частотой 1:4000 новорожденных. Тип наследования: аутосомно-доминантный. Ген нейрофиброматоза I типа расположен на длинном плече 17-й хромосомы. Описано более 100 типов мутаций. Наиболее частым признаком НФ I являются множественные светло-коричневые пятна типа «кофе с молоком». У 60% больных отдельные гиперпигментные пятна являются врожденными. С возрастом наблюдается тенденция к увеличению их числа. У детей в допубертатный период должно выявляться не менее 5 пятен с диаметром более 5 мм. Нейрофибромы являются диагностически важным признаком НФ I и могут появляться лишь в позднем детском возрасте или в юности. Опухолевые образования можно обнаружить по ходу нервных стволов, они не связаны с окружающими тканями, плотные на ощупь, 1-2 см в диаметре, безболезненные при пальпации. Другие диагностические признаки включают: пигментные пятна типа «веснушек» в кожных складках, узелки Лиша на радужной оболочке глаз, глиомы зрительного нерва, специфические костные дисплазии. Клинические проявления зависят от возраста больного, и поэтому для подтверждения диагноза часто необходимо наблюдать пациента на протяжении нескольких лет. Как правило, на первом году жизни у больных отмечаются только гиперпигментные пятна. Наличие у ребенка пораженного родственника первой степени родства позволяет поставить точный диагноз НФ I уже в этом возрасте. Течение заболевания прогрессирующее. Наиболее опасными проявлениями болезни Реклингхаузена являются опухоли, иногда из-за их злокачественности, иногда из-за места их расположения (черепно-мозговые нервы, малый таз, ЖКТ). Диагноз основан на совокупности клинических признаков, данных магнитно-резонансной томографии и молекулярно-генетического исследования. Дифференциальный диагноз проводится с другими заболеваниями из группы факоматозов. Многофакторные заболевания - докладчик Алена Многофакторные заболевания (мультифакториальные заболевания или болезни с наследственной предрасположенностью) - большая группа болезней (более 90%), развитие которых определяется взаимодействием определенных наследственных факторов (мутаций или сочетаний нормальных аллелей разных генов) и факторов среды. Болезни с наследственной предрасположенностью возникают у лиц с определенным генотипом (сочетание «предрасполагающих» аллелей) при провоцирующем действии факторов внешней среды. Наследственная предрасположенность к заболеванию может иметь моногенную или полигенную природу. Генетической основой моногенных форм наследственной предрасположенности являются мутации единичных генов, которые, как правило, наследуются по аутосомно-рецессивному (например, недостаточность лактазы) или Х-сцепленному рецессивному типу (например, недостаточность глюкозо-6-фосфатдегидрогеназы). Однако строго соответствующего законам Менделя распределения пораженных потомков в поколениях наблюдаться не будет. Это обусловлено тем, что для проявления данного патологического признака носитель мутации должен обязательно вступить в контакт со специфическим провоцирующим внешним фактором. В отсутствии подобного контакта мутантный ген остается «молчащим» и не проявляется в виде патологического признака. Определенными условиями среды для клинического проявления моногенных форм могут быть загрязнители воздушного бассейна, продукты питания, химический состав питьевой воды, различные ксенобиотики (лекарственные препараты) и т. д. Этиологической основой полигенных заболеваний с наследственной предрасположенностью является взаимодействие аллелей нескольких генов с комплексом внешнесредовых факторов. Соотношение влияния генетических и средовых факторов различно не только для данной болезни, каждого конкретного больного. При мультифакториальных заболеваниях, т.е. заболеваниях с наследственным предрасположением, основой оценки риска являются эмпирические данные о популяционной и семейной частоте каждого из них. Специфический генетический риск до 5% принято считать низким, до 10% —повышенным в легкой степени, до 20% —средним, выше 20% — высоким. Генетический риск средней степени расценивают как противопоказание к зачатию или показание к прерыванию уже имеющейся беременности. Возможность проведения пренатальной диагностики является определяющей для принятия положительного решения в отношении завершения беременности. 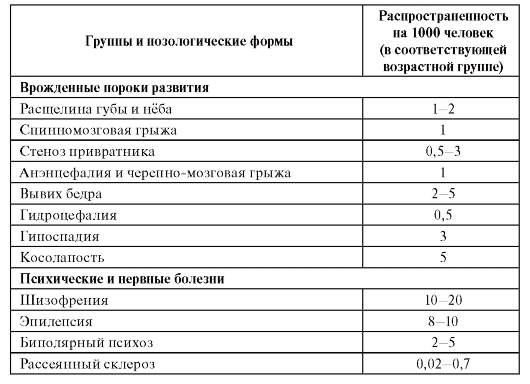 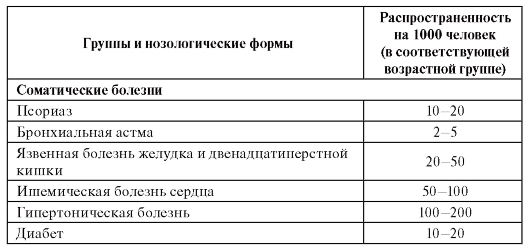 Частота широко распространенных мультифакториальных заболеваний: гипертоническая болезнь (10-20%), ИБС (5-10%), алкоголизм (1,4 - 10%), язвенная болезнь желудка и 12-перстной кишки (2-5%), сахарный диабет и шизофрения (1-2%). Примеры заболеваний Полигенная форма Гипертоническая болезнь - мультифакторное полиэтиологическое заболевание, характеризующееся повышением АД выше 140/90 мм рт.ст., с развитием симптомов поражения сердца, головного мозга, почек, сосудов при условии исключения вторичных артериальных гипертензий. Подверженность представлена: 1. Вклад генетических факторов – 69% 2. Материнским эффектом – 6% 3. Влияние среды – 25% Ишемическая болезнь сердца (ИБС) – это заболевание сердечно-сосудистой системы, связанное с нарушением равновесия между объемом коронарного кровообращения и метаболическими потребностями миокарда. Подверженность представлена: 1. Вклад генетических факторов – 52% 2. Влияние среды – 48% Язвенная болезнь желудка и ДПК - это мультифакториальное хроническое заболевание, сопровождающееся образованием язв в желудке с возможным прогрессированием и развитием осложнений. Подверженность представлена: 1. Вклад генетических факторов – 46% 2. Влияние среды – 54% 3. Материнский эффект – 0% Моногенная форма Наследственная непереносимость лактозы клинически проявляется повышением газообразования после приема молока или молочных продуктов. Частота встречаемости лактазной недостаточности сильно зависит от популяции. Частота в популяции: у европейцев - до 20%, у индейцев Америки - 70 -100%. В кишечнике взрослых гомозигот отсутствует b-галактозидаза, молочный сахар не расщепляется и не всасывается, брожение молочного сахара под действием кишечной флоры ведет к повышенному газобразованию. Недостаточность глюкозо-6-фосфатдегидрогеназы - распространенное наследственное заболевание, вызываемое дефектом фермента глюкозо-6-фосфатдегидрогеназы, который вырабатывает NADPH и защищает эритроциты от окислительного стресса. Спровоцировать могут лекарственные средства через 2-3 дня после приема: противомалярийные препараты, сульаниламиды, анальгетики, некоторые химиопрепараты, витамин К, растительные продукты (бобовые, стручковые). Частота встречаемости: в областях, эндемичных по малярии, имеет распространенность от 5-25%; в неэндемичных областях распространенность менее 0,5%. |