Проект анализ дротаверина гидрохлорид. Умарова К. проект дротаверина гидрохлорид. "Методы анализа лекарственных препаратов. Анализ дротаверина гидрохлорид"
 Скачать 71.11 Kb. Скачать 71.11 Kb.
|

|
Министерство образования и науки Республики Казахстан Карагандинский государственный университет имени академика Е. А. Букетова Химический факультет Кафедра физической и аналитической химии Проект по дисциплине: «Количественный химический анализ» на тему: “Методы анализа лекарственных препаратов. Анализ дротаверина гидрохлорид” Выполнила: студентка группы ТФП-20р п-я К. М. Умарова Руководитель: доцент кафедры физической и аналитической химии И. А. Пустолайкина Караганда 2020 Содержание Введение..................................................................................................................3 1. Основные принципы фармацевтического анализа....................................4 1.1. Классификация лекарственных средств ....................................................4-6 1.2. Система государственной регистрации и контроля качества лекарственных средств.......................................................................................7-9 1.3. Структура фармакопейной статьи...........................................................10-15 2. Требования к качеству дротаверина гидрохлорида................................16 2. 1. Общее описание фармацевтической субстанции (название, структурная формула, относительная молекулярная масса, свойства и т. п.).................16-17 2.2. Методы качественного определения дротаверина гидрохлорида.............18 2.3. Методы количественного определения дротаверина гидрохлорида...19-23 Выводы..................................................................................................................24 Список литературы.............................................................................................25 Введение Фармацевтический анализ – это наука о химической характеристике и измерения биологически активных веществ на всех этапах производства от контроля сырья до оценки качества полученного лекарственного вещества, изучения его стабильности, установления сроков годности и стандартизации готовой лекарственной формы. Фармацевтический анализ имеет свои специфические особенности, отличающие его от других видов анализа. Эти особенности заключаются в том, что анализу подвергают вещества различной химической природы: неорганические, элементоорганические, радиоактивные, органические соединения от простых алифатических до сложных природных биологически активных веществ. Чрезвычайно широк диапазон концентраций анализируемых веществ. Объектами фармацевтического анализа являются не только индивидуальные лекарственные вещества, но и смеси, содержащие различное число компонентов. Количество лекарственных средств с каждым годом увеличивается. Это вызывает необходимость разработки новых способов анализа. Лекарственное вещество — индивидуальное химическое соединение или биологическое вещество, обладающее лечебными или профилактическими свойствами. В настоящее время создается огромное количество лекарственных веществ, но также много и подделки. По данным Всемирной организации здравоохранения (ВОЗ), наибольший процент подделок приходится на антибиотики - 42%. В нашей стране, по информации Минздрава, фальсифицированные антибиотики составляют сегодня 47 % от общего числа препаратов – подделок, гормональные средства-1%,противогрибковые средства, анальгетики и препараты, влияющие на функцию желудочно -кишечного тракта -7%. Тема качества лекарственных препаратов всегда будет актуальна, так как от потребления этих веществ зависит наше здоровье, потому для дальнейших исследований мы взяли именно эти вещества. Цель исследования: познакомиться со свойствами лекарственных препаратов и установить их качество с помощью химического анализа. Задачи: - Изучить литературу (научную и медицинскую) с целью установления состава изучаемых лекарственных веществ, их классификации, химических, физических и фармацевтических свойств. - Подобрать методику, подходящую для установления качества выбранных лекарственных препаратов в аналитической лаборатории. - Провести исследование качества лекарственных препаратов по выбранной методике качественного анализа. - Проанализировать результаты, обработать их и оформить работу. Основные принципы фармацевтического анализа Классификация лекарственных средств Классификация огромного арсенала ЛС (в РБ зарегистрировано свыше 6000 наименований) имеет большое значение не только для создания рациональной системы информации о ЛС, но и проведения исследований по созданию новых ЛВ. Любая классификация не может быть постоянной. Создание новых ЛС, достижения в области фармакологии требуют постоянного совершенствования классификации ЛС. Существуют два основных типа классификации ЛС: химическая – по химической структуре содержащегося лекарственного вещества и фармакологическая – по характеру действия ЛС на организм. Каждая из этих классификаций имеет свои положительные и отрицательные стороны. Фармакологическая классификация отражает принципы преимущественного действия ЛВ на ту или иную физиологическую систему (сердечно-сосудистую, центральную нервную и т.д.). Однако в одну и ту же группу при этом попадают ЛС, различные по химическому строению. Химическая классификация позволяет четко распределить все ЛС по группам и классам соединений в соответствии с их химической структурой. Но зачастую в одной и той же группе оказываются ЛС с различным фармакологическим действием. Она также имеет важное значение для проведения исследований в области синтеза, получения ЛС из растительного и животного сырья, установления связи между их химической структурой и фармакологическим действием. Все ЛВ в соответствии с химической классификацией подразделены на две большие группы: неорганические и органические. Неорганические классифицируют в соответствии с положением элементов в Периодической системе Д.И. Менделеева и по основным классам: оксиды, гидроксиды, соли, комплексные соединения. Органические ЛВ классифицируют аналогично тому, как это принято в органической химии. При этом используют два классификационных признака: структуру углеродной цепи или цикла и природу функциональной группы. По первому признаку органические ЛВ подразделают на алифатические (ациклические) и циклические, последние в свою очередь – на карбоциклические и гетероциклические соединения. Карбоциклические соединения объединяют два ряда веществ – алициклические и ароматические. Органические ЛВ, структура которых включает только атомы углерода и водорода (углеводороды), классифицируют как производные углеводородов, в молекуле которых один или несколько атомов водорода замещены на функциональные группы. По второму классификационному признаку в зависимости от наличия в молекуле той или иной функциональной группы алифатические и ароматические углеводороды подразделяют на галогенопроизводные, спирты, фенолы, простые и сложные эфиры, альдегиды и их производные (амины, оксимы, гидразоны, семикарбазоны, тиосемикарбазоны), кетоны, сульфокислоты, карбоновые кислоты и их производные (соли, ангдириды, амиды, гидразиды и др.), нитро- и нитрозосоединения, амины, гидразины и азосоединения. Гетероциклические соединения классифицируют по числу атомов, образующих цикл, природе гетероатомов и их количеству, а также по числу гетероциклов или характеру конденсированной системы, включающей гетероциклы и ароматические циклы. Разобраться в применении огромного многообразия арсенала современных ЛС может помочь фармакотерапевтическая классификация. В соответствии с существующими требованиями, ЛС в ней должны распределяться по классам, затем по входящим в каждый из них группам и подгруппам. Каждое ЛС с его основным названием и синонимами должно иметь в подгруппах точную локализацию. В 1996 году ВОЗ опубликовали предложенный вариант «Анатомо-терапевтическо-химической классификации лекарственных субстанций» (АТХ). По ней все ЛС классифицируются на 14 групп в зависимости от органа или системы, на который они действуют. Каждая группа включает терапевтические и фармакологические, а в некоторых случаях химические подгруппы. Каждая субстанция и лекарственная форма имеет свой буквенный и цифровой индекс. Таким образом, АТХ представляет собой «банк данных», в котором чётко индексировано каждое ЛС. Этот классификационный документ рассчитан, в первую очередь, на использование органами здравоохранения при планироывнии лекарственного обеспечения населения. Для использования врачами и провизорами АТХ представляет значительную сложность. Широкое признание у врачей и провизоров получила фармакотерапевтическая классификация, разработанная профессором М. Д. Машковским, в наиболее современном виде представления в последних изданиях пособия для врачей «Лекарственные средства». Она помогает установить, к какой группе относится ЛС, является ли оно новым или аналогом существующих, каковы его синонимы, состав. По этой классификации ЛС распределены по характеру действия на системы, органы, процессы, по 13 основным классам. Эти классы разделены на группы, а последние – на подгруппы, исходя из следующих признаков: основные фармакологические свойства, основные области медицинского применения, сходство в химической структуре. Это даёт представление о существовании связи между химической структурой и фармакологическим действием субстанций. В каждой группе (подгруппе) первыми представлены ЛС - «родовые препараты», характеризующие основные черты данной группы. Описание остальных дополняет и развивает представление о группе в целом. Менеджеры оптовых фирм-поставщиков ЛС используют алфавитную классификацию. Все ЛС вносятся в прайс-лист по алфавиту под торговыми наименованиями, в прайс-листе присутствует информация о форме выпуска ЛС и его дозировке, производителе, сроке годности, приводится МНН. В контрольно-аналитических лабораториях также используется алфавитный каталог (торговые наименования) нормативной документации по контролю качества ЛС. Система государственной регистрации и контроля качества лекарственных средств Государственная регистрация – система учета, допуска к промышленному производству, реализации и медицинскому применению лекарственных средств (ЛС), изделий медицинского назначения и медицинской техники, производимых в Республики Казахстан или требованиям по качеству, эффективности и безопасности для человека, установленным в РК. Государственная регистрация лекарственных средств и фармацевтических субстанций включает: - прием РУП «Центр экспертиз и испытаний в здравоохранении» (РУП «ЦЭИЗ») регистрационного досье, включающего в себя документы, необходимые для проведения государственной регистрации лекарственных средств и фармацевтических субстанций; - заключение между юридическим лицом, производящим ЛС, либо размещающим заказ на промышленное производство в иной организации, либо входящим в состав объединения, в которое также входит производитель ЛС, в РУП «ЦЭИЗ» договора на оказание услуг по организаций и проведению работ для государственной регистрации (перерегистрации) ЛС и фармацевтических субстанций; - проведение в государственных организациях здравоохранения биоэквивалентных испытаний генерических ЛС, клинических испытаний оригинальных ЛС, впервые заявленных для государственной регистрации в РБ или ранее зарегистрированных ЛС, но предлагаемых для применения по новым медицинским показаниям; - проведение в государственных организациях здравоохранения апробации методик анализа ЛС и фармацевтических субстанций, впервые заявленных для государственной регистраций в РБ и контроля за качеством ЛС при назначении их клинических испытаний; - проведение работниками МЗ РК инспекционной проверки порядка осуществления промыленного производства зарубежным производителем ЛС, который впервые подает заявку для государственной регистрации в РК; - принятие МЗ РК решения о государственной регистрации (отказе в государственной регистрации) ЛС и фармацевтических субстанций. Комплекс работ по регистрации или перерегистрации ЛС, а также ведение Государственного реестра осуществляется РУП «Центр экспертиз и испытаний в здравоохранении». Данная организация проводит экспертизу регистрационного досье на ЛС, включающего в себя документы по качеству (фармакопейная статья или другая аналитическая документация), эффективности и безопасности ЛС (данные о проведении клинических испытаний оригинальных ЛС, проведении биоэквивалентных испытаний генерических ЛС). При необходимости методики контроля качества, поданных на регистрацию лекарственных средств, апробируются в Лаборатории Фармакопейного и фармацевтического анализа РУП «ЦЭИЗ» или других государственных организациях Министерства здравоохранения. Документы по эффективности и безопасности ЛС рассматриваются в Республиканской клинико-фармакологической лаборатории РУП «ЦЭИЗ». Вся экспертиза регистрационного досье обычно продолжается не более 30 дней. В случае государственной регистрации оригинальных ЛС, назначении клинических испытаний ЛС, возникновении разногласий между экспертами или несогласия заявителя с замечаниями экспертов экспертные заключения о регистрации ЛС дополнительно рассматриваются комиссией по лекарственным средствам. После проведения экспертизы РУП «ЦЭИЗ» представляет проект решения о государственной регистрации ЛС в Министерство здравоохранения, которое и принимает окончательное решение о государственной регистрации ЛС и внесении его в Государственный реестр лекарственных средств, разрешенных к промышленному производству и применению на территории РК. Система контроля качества ЛС. Высокое качество производимых и экспортируемых в Республику Казахстан лекарственных средств обеспечивается лабораториями, осуществляющими контролирующие функции. Данные лаборатории подразделяются на: - осуществляющие функцию контроля качества ЛС на фармацевтических предприятиях (отделы контроля качества предприятий проверяют качество ЛС по всем показателям, указанным в нормативной документации); - осуществляющие функции государственного контроля качества ЛС (Республиканская контрольно-аналитическая лаборатория РУП «ЦЭИЗ» и другие аккредитованные испытательные лаборатории (ИЛ)). Порядок проведения контроля качества ЛС определяется соответсвующими документами. В настоящее время действует Постановление МЗ РК №55 от 14 февраля 2005 года «Инструкция по осуществлению государственного надзора за безопасностью, эффективностью и качеством лекарственных средств». В этом постановлении определен перечень документов по качеству ЛС, представляемым в ИЛ (сертификат качеств или аналитический паспорт предприятия-изготовителя); порядое передачи и хранения образцов; объемы и сроки проводимых фармацевтических анализов (например, лекарственные средства для внутреннего употребления с маркировкой на упаковке: «Детское» или при наличии указания в инструкции по медицинскому применению об использовании для детей в возрасте до двух лет проверяются по показателям: «Описание», «Подлинность», «Упаковка», «Маркировка»).При проведении контроля качества ЛС используются следующие нормативные документы:- нормативные документы по контролю качества лекарственных средств зарубежного производства, разрешенные для использования Министерством здравохранения Республики Казахстан;- Государственная Фармакопея РК и фармакопейные статьи на лекарственные средства отечественного производства, утвержденные Министерством здравоохранения Республики Казахстан.Государственная Фармакопея РК составлена на основе Европейской Фармакопеи, в ее структуру входят следующие разделы: общие сведения; методы анализа; реактивы; общие и частные статьи на лекарственные формы и субстанции; частные статьи на лекарственное растительное сырье и др.Структура фармакопейной статьи Государственная фармакопея (ГФ) – сборник общих и частных фармакопейных статей, устанавливающих требования к качеству лекарственных средств, лекарственного растительного сырья, фармацевтических субстанций и вспомогательных веществ. Основу Государственной фармакопеи составляют общие и частные фармакопейные статьи. Общие фармакопейные статьи описывает принятые в фармакопейном анализе общие положения, методы анализа или включают в себя перечень нормируемых показателей и методов испытаний определенной лекарственной формы. Частные фармакопейные статьи определяет уровень требований к конкретным лекарственным средствам. Фармакопейная статья (ФС) – технический нормативный правовой акт, документ, устанавливающий требования к качеству лекарственных средств, фармацевтических субстанций, лекарственного растительного сырья, вспомогательных веществ, реактивов, упаковочных материалов, используемых в промышленном производстве, аптечном изготовлении лекарственных средств, к стандартным образцам, используемым при проверке качества ЛС, методам контроля за качеством, их упаковке, условиям и срокам хранения. ФСП – фармакопейная статья предприятия (разрабатывается на лекарственную форму, выпускаемую заводом из субстанций, соответсвующим требованиям ФС). Названия. Кроме названий на русском языке, приводится также латинское название. Это название может использоваться вместо русского названия, равно как и любой другой синоним, который признан эквивалентным компонентным уполномоченным органом. Относительные атомные и молекулярные массы. Относительная атомная масса (А.м.) или относительна молекулярная масса (М.м.) указываются, когда это необходимо, в начале частной фармакопейной статьи. Относительную атомную массу, относительную молекулярную массу, молекулярную массу и графическую формулу приводят как информативный материал. Вводная часть фармакопейных статей. В вводной части, удищей после названия монографии, приводится официальное определение субстанции, готового лекарственного средства или иного продукта, являющегося предметом частной фармакопейной статьи. Пределы содержания. Если указаны пределы содержания, то это пределы полученные с использованием метода, указанного в разделе «Количественное определение». Лекарственные средства, содержащие лекарственное растительное сырье. В частных статьях на лекарственные средства, содержащие лекарственное растительное сырье, вводная часть включает указание на предмет частной статьи. Это может быть, например, лекарственное растительное сырье в исходном виде или лекарственное растительное сырье, измельченное в порошок. Если частная статья распространяется на несколько вариантов, например, на оба из указанных, то это оговаривается во вводной части. Производство. Информация в разделе «Производство» призвана привлечь внимание к некоторым важным аспектам процесса производства и не обязательно является исчерпывающей. Содержащиеся в ней инструкции адресованы производителю. Они могут относиться, например, к материалам, к процессу производства, к его валидации и контролю, к постадийному контролю, а также к испытаниям, которые производитель должен проводить перед выпуском для каждой серии продукта или для выбранных серий. Эти положения обязательно должны быть потверждены посредством анализа конечного продукта. Компетентным уполномоченным органом может быть установлено, что приведенные вышеуказанные аспекты были выполнены. Такое заключение может быть сделано на основании проверки полученных от производителя данных, или при инспектировании производства, или при испытании соответсвующих образцов. Отсутствие раздела «Производство» не означает, что аспекты процесса производства, отмеченные выше, не требуют внимания. Любой описанный в частной фармакопейной статье продукт должен производиться в соответствии с принципами надлежащей производственной практики (GMP) и соответствующими международными соглашениями, а также национальными и наднациональными законами, распространяющимися на продукты, предназначенные для использования в медицине. В разделе «Производство» в частной фармакопейной статье на вакцину могут быть указаны свойства штамма и тестовые методы для подтверждения этих свойств. Эти методы приводятся для информации в качестве примера. Описание (свойства). Указывают характеристики физического состояния и цвет лекарственного средства; если необходимо, приводят информацию о запахе и гигроскопичности. Твердые субстанции могут быть крупнокристаллическими (не более 40% частиц порошка должно быть размером менее 0,4 мм), кристаллическими (не менее 95% частиц порошка должно быть размером менее 0,4 мм и не более 40% - размером менее 0,2 мм), мелкокристаллическими (не менее 95% частиц порошка должно быть размером менее 0,2 мм) или аморфными (при вращении столика микроскопа не наблюдается отражение света). Характеристики кристалличности и гигроскопичности в описании приводятся для информации и испытанию не подлежат. При необходимости нормирования величины частиц в частной фармакопейной статье приводят специальный раздел. Цвет характеризуется названиями: белый, желтый, оранжевый, красный и др. При оттеночных цветах на первом месте указывают тот цвет, который содержится в меньшей доле, а затем через дефис – преобладающий цвет (например, красно-коричневый). Слабоокрашенные образцы имеют оттенок цвета, название которого характеризуют суффиксом «-оват» (например, «желтоватый») или указывают «светло-» (например, «светло-желтый»). Цвет твердых веществ определяются на матово-белом фоне (белая плотная или фильтровальная бумага) при рассеянном дневном свете в условиях минимального проявления тени. Небольшое количество вещества помещают на белую бумагу и без нажима равномерно распределяют по поверхности бумаги (осторожно разравнивают шпателем или другим приспособлением) так, чтобы поверхность оставалась плоской. Запах. Запах характеризуется терминами: «без запаха», «с характерным запахом», «со слабым характерным запахом». В случае легко летучих жидкостей наносят 0,5 мл на фильтровальную бумагу и запах определяют сразу же после нанесения, если нет других указаний в частной фармакопейной статье. Растворимость. Длят определения растворимости используют растворители, охватывающие широкую шкалу полярности (вода...спирт...ацетон...гексан). Для характеристики растворимости используют количество (мл) растворителя, необходимое для растворения 1 г вещества. - очень легко растворим до 1; - легко растворим от 1 до 10; - растворим от 10 до 30; - умеренно растворим от 30 до 100; - мало растворим от 100 до 1000; - очень мало растворим от 1000 до 10000; - практически нерастворим более 10000; - частично растворим. Термин используется для характеристики смесей, содержащих как растворимые, так и нерастворимые компоненты; - смешивается с.... Термин используется для характеристики жидкостей, смешивающихся с указанным растворителем во всех соотношениях. Для определения растворимости навеску вещества вносят в отмеренное количество растворителя и непрерывно встряхивают в течение 10 минут при температуре 20±5ºС. Предварительно образец может быть растерт. Для медленно растворимых образцов, требующих для своего растворения более 10 минут, допускается также нагревание на водяной бане до 30ºС; наблюдение производят после охлаждения раствора до температура 20±5ºС и энергичного встряхивания в течение 1-2 минут. Вещество считают растворившимся, если в растворе при наблюдении в проходящем свете не обнаруживаются частицы вещества. Для веществ, образующих при растворении мутные растворы, соответсвующее указание должно быть приведено в частной фармакопейной статье. Если указано, что субстанция растворима в жирных маслах, то имеется в виду, что она растворима в любом масле, относящемуся к классу жирных масел. Подлинность (идентификация). Для установления подлинности субстанции обычно используются инструментальные методы анализа (чаще всего ИК-спектроскопия) в сочетании с химическими (качественные реакции на функциональные группы). Обычно для определения подлинности фармакопейная статья предлагает 3-5 испытаний. В некоторых частных фармакопейных статьях имеются подразделы «Первая идентификация» и «Вторая идентификация». Обычно используют только первую идентификацию. Температура плавления. Испытание обычно применяют для характеристики твердых веществ. Температура затвердевания; температура кипения (температруные пределы перегонки); плотность; вязкость; показатель преломления. Данные испытания вводят для характеристики жидких субстанций. Удельное вращение. Вводят для характеристики оптически активных веществ. Удельный показатель поглощения. Данный показатель является дополнительной характеристикой подлинности и чистоты субстанции. Прозрачность и цветность раствора. Данные испытания обязательно проводят для субстанций и лекарственных средств, предназначенных для парентерального введения. Испытания особенно актуальны для ЛС, изменяющих свой цвет при окислении кислородом воздуха. рН (кислотность или щелочность). Для проведения данного испытания используется потенциометрическое измерение значения рН. Допустимый интервал рН обычно должен быть не более 2 (например, значение рН должно быть от 5,5 до 7,5). Испытания проводят для субстанций и лекарственных средств, предназначенных для парентерального введения. Примеси. Данное испытание предусматривает контроль продуктов деструкции и технологических примесей (полупродуктов и побочных продуктов синтеза), так называемые «сопутствующие примеси». При этом могут контролироваться как «охарактеризованные примеси» (т.е. те, которые ранее были признаны компетентными органами в качестве охарактеризованных; туда могут также быть включены и примеси, которые считаются охарактеризованными другими способами, например, примеси, которые встречаются в виде естественных метаболитов) и «другие определяемые примеси» (например, потенциальные примеси, которые не были определены в каких-либо образцах субстанции во время разработки монографии или которые встречаются в концентрациях менее 0,1%, но содержание которых может ограничиваться с помощью тестов, описываемых в частной фармакопейной статье). Для определения примесей чаще всего используются различные хроматографические методы анализа. Неорганические анионы (хлориды, сульфаты и др.). Выбор контролируемых анионов определяется технологией получения субстанции. При этом контролируемые анионы могут быть нетоксичными (например, хлориды, сульфаты и т.д.). Контроль анионов не вводят, если они входят в состав субстанции (например, вещество является гидрохлоридом или сульфатом). Неорганические катионы (железо, медь и др.). Это испытание вводят, если контроль содержания отдельных катионов является существенным для качества субстанции; их содержание должно быть обосновано. Контроль катионов не вводят, если они входят в состав субстанции (например, железа лактат). Сульфатная зола. Как правило, сульфатная зола не должны превышать 0,1%. Отсутсвие этого испытания в частной фармакопейной статье или повышенное содержание сульфатной золы требует соотвествующего обоснования. Тяжелые металлы. Содержание тяжелых металлов не должно превышать 0,001%, если нет других указаний в частной фармакопейной статье. Мышьяк. Данное испытание вводят в том случае, когда или исходное сырье может содержать мышьяк, например, для сырья природного происхождения, или возможно загрязнение им в процессе получения субстанции. Содержание мышьяка, как правило, не должно превышать 0,0001%. Остаточные органические растворители. Содержание остаточных количеств органических растворителей, использующихся при получении субстанции, должно соответствовать требованиям ФС. Бактериальные эндотоксины (пирогенность). Данные испытания приводят для субстанций, предназначенных для приготовления лекарственных ср,едств для парентерального применения. Указанные субстанции должны выдерживать тест на бактериальные эндотоксины или пирогенность без проведения предварительной стерилизации. Микробиологическая чистота. Микробиологическая чистота лекарственных средств должна соответствовать требованиям ГФ РК общих монографий 5.1.8. Микробиологическая чистота лекарственных растительных препаратов для орального применения или 5.1.4. Микробиологическая чистота нестерильных лекарственных препаратов и субстанций для фармацевтического применения . Стерильность. Данное испытание вводят для субстанций, используемых в производстве готовых стерильных лекарственных средств, которые не подвергаются процедуре стерилизации. Потеря в массе при высушивания. Испытание вводят для контроля содержания летучих веществ и/или влаги в субстанции. Введение одного из этих испытаний в ФС, как правило, обязательно. Отсутствие их должно быть обосновано. Если нет других указаний в частной фармакопейной статье и субстанция не является кристаллогидратом (кристаллосольватом), потеря в массе при высушивании или содержание воды не должно превышать 0,5%. Если субстанция является кристаллогидратом (кристалосольватом), регламентируют верхний и нижний пределы. Количественное определение. Для количественного определения активного вещества субстанции или лекарственной формы используют инструментальные и химические методы анализа. Хранение. Упаковка и условия хранения должны обеспечивать качество лекарственного средства в течение установленного срока годности. Ниже приводится расшифровка рекомендуемых температурных условий хранения препаратов:
Маркировка. Маркировка регламентируется компетентным уполномоченным органом с изданием соответсвующего нормативного правового акта. Таким образом, информация в разделе «Маркировка» не претендует на полноту. Она ориентирована прежде всего на фармакопейные цели, и обязательными являются только те положения, которые необходимы для подтверждения соответствия продукта статье. Вся остальная информация носит рекомендательный характер. В тех случаях, когда в Фармакопее употребляется термин «этикетка», соответствующая информация может быть помещена на контейнере, на упаковке или во вкладыше, в зависимости от решения компетентного уполномоченного органа. Требования к качеству дротаверина гидрохлорида Общее описание фармацевтической субстанции (название, структурная формула, относительная молекулярная масса, свойства и т. п.) Структурная формула 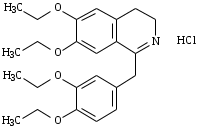 Русское название Дротаверина гидрохлорид Латинское название вещества Дротаверина гидрохлорид Drotaverinum hydrochloricum Химическое название 1-[(3,4-Диэтоксифенил)метил]-6,7-диэтокси-3,4-дигидроизохинолина гидрохлорид Брутто-формула C24H31NO4 • HCl Молярная масса 434,0 Cодержит не менее 98,0 % и не более 101,0 % дротаверина гидрохлорида C24H31NO4 • HCl в пересчете на сухое вещество. Описание. От светло-желтого до зеленовато-желтого цвета кристаллический порошок почти без запаха. Растворимость. Легко растворим в хлороформе, растворим в спирте 96 %, умеренно растворим в воде. Фармакологическое действие. Сосудорасширяющее, спазмолитическое, миотропное, гипотензивное. Хранение. В защищенном от света месте при температуре не выше 25 °С. Методы качественного определения дротаверина гидрохлорида Дротаверина гидрохлорид один из наиболее известных и широко применяемых в медицинской практике миотропных спазмолитических средств. Для количественного определения дротаверина гидрохлорида в субстанциях и сложных лекарственных формах предлагаются различные титрометрические и физико-химические методы анализа. Применение титрометрических методов анализа для контроля качества лекарственных средств является трудоемким и длительным, требует предварительной стандартизации титранта. Хроматографические и спектрофотометрические методы эффективны, но дороги, требуют раствора стандартного образца. 1. Качественная реакция. 10 мг субстанции растворяют в 5 мл серной кислоты концентрированной, прибавляют 30 мкл 0,1 М раствора железа(III) хлорида; при нагревании смеси должно появиться зеленое окрашивание. После охлаждения прибавляют 30 мкл азотной кислоты разведенной 16 %; должно появиться коричнево-красное окрашивание. 2. Качественная реакция. 50 мг субстанции растворяют в 2 мл спирта 96 %; полученный раствор должен давать характерную реакцию на хлориды (ОФС «Общие реакции на подлинность»). Методы количественного определения дротаверина гидрохлорида 1. ИК-спектр. Инфракрасный спектр субстанции, снятый в диске с калия бромидом, в области от 4000 до 400 см-1 по положению полос поглощения должен соответствовать спектру стандартного образца дротаверина гидрохлорида. 2. УФ-спектр. Ультрафиолетовый спектр 0,0015 % раствора субстанции в 0,1 М растворе хлористоводородной кислоты в области длин волн от 210 до 440 нм должен иметь максимумы поглощения при 241 ± 2 нм, 302 ± 2 нм и 353 ± 2 нм и минимумы поглощения при 223 ± 2 нм, 262 ± 2 нм и 322 ± 2 нм. Температура плавления. От 208 до 212 ºС (с разложением, ОФС «Температура плавления», метод 1, прибор 2). Прозрачность раствора. Раствор 0,1 г субстанции в 10 мл воды должен быть прозрачным (ОФС «Прозрачность и степень мутности жидкостей»). Цветность раствора. Окраска раствора, полученного в испытании «Прозрачность раствора», должна выдерживать сравнение с эталоном GY3, но быть интенсивнее окраски эталона GY4 (ОФС «Степень окраски жидкостей»). рН. От 3,5 до 5,5 (1 % раствор, ОФС «Ионометрия», метод 3). Родственные примеси. Определение проводят методом ВЭЖХ. Подвижная фаза (ПФ). Метанол – ацетонитрил – ацетатный буферный раствор 8:48:44. Ацетатный буферный раствор. 2,176 г натрия ацетата безводного растворяют в 500 мл воды, прибавляют 6 мл уксусной кислоты ледяной, перемешивают, доводят объем раствора водой до 1000 мл и перемешивают. Испытуемый раствор. Около 10 мг (точная навеска) субстанции растворяют в 3 мл ПФ и доводят тем же растворителем до 5,0 мл. Раствор сравнения А. 0,5 мл испытуемого раствора доводят ПФ до 100,0 мл. Раствор сравнения Б. 20 мг субстанции растворяют в 19 мл воды при нагревании до 40-50 ºС, прибавляют 5 мг растертого калия перманганата, перемешивают в течение 2 мин, прибавляют 1 мл фосфорной кислоты концентрированной и вновь перемешивают до получения прозрачного раствора. Хроматографические условия
Хроматографируют испытуемый раствор и растворы сравнения А и Б. Пригодность хроматографической системы. На хроматограмме раствора сравнения Б: - после пика дротаверина должны элюироваться 3 пика продуктов окисления; - разрешение (R) между соседними пиками должно быть не менее 3,0. Допустимое содержание примесей. На хроматограмме испытуемого раствора: - площадь пика любой примеси должна быть не более площади основного пика на хроматограмме раствора сравнения А (не более 0,5 %); - суммарная площадь пиков всех примесей не должна более чем в 2 раза превышать площадь основного пика на хроматограмме раствора сравнения А (не более 1,0 %). Не учитывают пики, площадь которых менее 10 % от площади основного пика на хроматограмме раствора сравнения А (менее 0,05 %). Вода. Не более 3,0 % (ОФС «Определение воды», метод 1). Около 0,5 г (точная навеска) субстанции растворяют в 10 мл смеси хлороформ – метанол 9:1 и титруют реактивом К.Фишера. Для данной цели пригодны инструментальные или микроаналитические методы. При использовании обычных количеств (около 50 мг стандартного образца) титрование выполняют реагентом, разбавленным в 2-5 раз. Сульфатная зола. Не более 0,1 % (ОФС «Сульфатная зола»). Для определения используют около 1 г (точная навеска) субстанции. Тяжелые металлы. Не более 0,002 %. Определение проводят в соответствии с требованиями ОФС «Тяжёлые металлы», метод 2, в зольном остатке, полученном после сжигания 1 г субстанции (ОФС «Сульфатная зола»), с использованием эталонного раствора 2. Остаточные органические растворители. В соответствии с ОФС «Остаточные органические растворители». *Бактериальные эндотоксины. Не более 4,3 ЕЭ на 1 мг субстанции (ОФС «Бактериальные эндотоксины»). Для проведения испытания готовят исходный раствор субстанции (концентрация 10 мг/мл), а затем разбавляют его не менее чем в 200 раз. Микробиологическая чистота. В соответствии с ОФС «Микробиологическая чистота». Количественное определение. Около 0,3 г (точная навеска) субстанции растворяют в 20 мл уксусной кислоты ледяной, прибавляют 3 мл раствора ртути(II) ацетата и титруют 0,1 М раствором хлорной кислоты до появления зеленого окрашивания (индикатор - 0,1 % раствор кристаллического фиолетового). Параллельно проводят контрольный опыт. 1 мл 0,1 М раствора хлорной кислоты соответствует 43,40 мг дротаверина гидрохлорида С24Н31NO4 • HCl. Спектрофотометрическое определение дротаверина гидрохлорида Для спектрофотометричекого определения дротаверина гидрохлорида в качестве образца сравнения был выбран калия дихромат, т.к. аналитическая длина волны дротаверина (353 нм) входит в интервал, оптимальный для калия дихромата (340,5–359,5 нм). Для определения содержания дротаверина в субстанции по калию дихромату авторами вводится коэффициент пересчета: 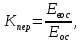 где 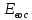 – удельный показатель поглощения внешнего образца сравнения, – удельный показатель поглощения внешнего образца сравнения, 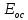 – удельный показатель поглощения определяемого вещества. – удельный показатель поглощения определяемого вещества. Коэффициент пересчета для калия дихромата равен 0,434. В данной работе в качестве объектов исследования были выбраны лекарственные формы, основным действующим веществом которых является дротаверина гидрохлорид: 1) Дротаверин (ОАО «Органика», г. Новокузнецк, Россия); 2) Дротаверин (ОАО «Ирбитский химико-фармацевтический завод», г. Ирбит, Россия); 3) Дротаверин (ЗАО «Производственная фармацевтическая компания Обновление», г. Новосибирск, Россия); 4) Ношпа (ЗАО «Хиноин», Венгрия); 5) Пенталгин (ОАО «Фармстандарт-Лексредства», г. Курск, Россия). Методика количественного определения дротаверина гидрохлорида в субстанции спектрофотометрическим методом: Готовят раствор образца сравнения калия дихромата для анализа дротаверина. Для этого точную массу калия дихромата (0,1 г) помещают в мерную колбу вместимостью 50 мл, доводят объем до метки дистилированной водой и перемешивают. 1 мл полученного раствора помещают в мерную колбу вместимостью 50 мл, доводят объем до метки 0,1М раствором хлористоводородной кислоты и перемешивают. Для приготовления раствора исследуемой субствнции таблетку, содержащую дротаверин, взвешивают на аналитических весах с погрешностью 0,0001 г, затем ее измельчают, переносят в мерную колбу на 50 мл и доводят объем до метки дистилированной водой и тщательно перемешивают. 0,5 мл полученного раствора помещают в мерную колбу вместимостью 50 мл, доводят объем раствора 0,1М раствором хлористоводородной кислоты до метки и перемешивают. Измеряют оптическую плотность полученного раствора дротаверина и калия дихромата при длине волны 353 нм относительно 0,1М раствора хлористоводородной кислоты. Расчет результатов количественного определения дротаверина гидрохлорида проводят по формуле: 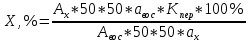 где, 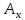 и и 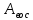 – оптические плотности определяемого вещества и образца сравнения соответственно; – оптические плотности определяемого вещества и образца сравнения соответственно;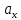 и и 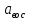 – точные навески определяемого вещества и образца сравнения соответственно; – точные навески определяемого вещества и образца сравнения соответственно; Во всех проверенных образцах содержание дротаверина гидрохлорида соответствует норме и лишь немного отличается от заявленного содержания на упаковке. Выводы 1. Анализ литературы показал, что для идентификации дротаверина гидрохлорид предложены спектрофотометрические методики, для обнаружения посторонних примесей в субстанциях дротаверина – хроматографические методы. 2. Гармонизованы требования к качеству субстанций и однокомпонентных таблетированных лекарственных форм дротаверина гидрохлорида по показателям, в нормировании которых были отмечены разночтения (прозрачность раствора дротаверина гидрохлорида, цветность раствора, растворимость субстанции, температура плавления дротаверина гидрохлорида и др.) 3. Предложены методики подтверждения подлинности дротаверина гидрохлорида в субстанциях и однокомпонентных таблетках. 4. Предложены методики определения посторонних примесей в субстанциях и однокомпонентных таблетках дротаверина гидрохлорида. 5. Разработана методика определения дротаверина гидрохлорида методом спектрофотометрии с дихроматом калия в диапазонах ее концентрации 0,8-1,0 мкМ, 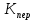 =0.434. =0.434.Список использованной литературы 1. Министерство здравоохранения Республики Казахстан.Государственная Фармакопея Республики Казахстан // первое издание, Т. 3 2. Д. В. Моисеев, В. А. Куликов. Учебное пособие «Фармакопейный анализ» // 2011. 3. Г.К. Зиятдинова. Количественное определение производных бензилизохинолина методом кулонометрического титрования / Г.К. Зиятдинова, А.И. Самигуллин, С.Г. Абдуллина, Г.К. Будников // Химико-фармацевтический журнал, 2008.– Т.42.– №2.– С.47–50. 4. Е.А. Илларионова. Новый вариант спектрофотометрического определения дротаверина / Е.А. Илларионова, И.П. Сыроватский, П.О. Иноземцев // Сибирский медицинский журнал, 2011.– №5.– 75–77. 5. Р. Кельнер. Аналитическая химия. Проблемы и подходы. В 2 т.: пер с англ.// М.: Мир, АСТ, 2004. |