зайко. Н. Н. Зайко Патологическая физиология Введение Предмет и задачи патологической физиологии Патологическая физиология есть наука, изучающая жизнедеятельность больного организма. Программа
 Скачать 7.32 Mb. Скачать 7.32 Mb.
|

|
С учетом изложенных представлений механизм начального этапа атеросклероза, характеризующегося избыточным накоплением липидов в интиме артерий, может быть обусловлен: 1. Генетической аномалией рецептор-опосредованного эндоцитоза ЛПНП (отсутствие рецепторов – менее 2% от нормы, уменьшение их числа – 2 – 30% от нормы). Наличие таких дефектов обнаружено при семейной гиперхолестеринемии (гипербеталипопротеидемия II А типа) у гомо- и гетерозигот. Выведена линия кроликов (Ватанабе) с наследственным дефектом рецепторов к ЛПНП. 2. Перегрузкой рецепторопосредованного эндоцитоза при алиментарной гиперхолестеринемии. И в том, и в другом случае наступает резкое усиление нерегулируемого эндоцитозного захвата частиц ЛП эндотелиальными клетками, макрофагами и гладкомышечными клетками стенки сосудов вследствие выраженной гиперхолестеринемии. 3. Замедлением удаления атерогенных липопротеидов из стенки сосудов через лимфатическую систему в связи с гиперплазией, гипертензией, воспалительными изменениями. Существенный дополнительный момент – различные превращения (модификации) липопротеидов в крови и сосудистой стенке. Речь идет об образовании в условиях гиперхолестеринемии аутоиммунных комплексов ЛП – IgG в крови, растворимых и нерастворимых комплексов ЛП с гликозаминогликанами, фибронектином, коллагеном и эластином в сосудистой стенке (А. Н. Климов, В. А. Нагорнев). По сравнению с нативными ЛП захват модифицированных ЛП клетками интимы, в первую очередь макрофагами (с помощью нерегулируемых холестерином рецепторов), резко возрастает. Это, как полагают, является причиной превращения макрофагов в так называемые пенистые клетки, которые составляют морфологическую основу стадии липидных пятен и при дальнейшем прогрессировании -атером. Миграция кровяных макрофагов в интиму обеспечивается с помощью моноцитарного хемотаксического фактора, образующегося под действием ЛП и интерлейкина-1, который выделяется из самих моноцитов. На заключительном этапе формируются фиброзные бляшки как ответ гладкомышечных клеток, фибробластов и макрофагов на повреждение, стимулируемый факторами роста тромбоцитов, эндотелиоцитов и гладкомышечных клеток, а также стадия осложненных поражений – кальцификация,тромбообразование и др. (рис. 19.13).  Приведенные выше концепции патогенеза атеросклероза имеют свои сильные и слабые стороны. Наиболее ценным достоинством концепции общих метаболических нарушений в организме и первичного липоидоза артериальной стенки является наличие экспериментальной холестериновой модели. Концепция первичного значения местных изменений в артериальной стенке, несмотря на то что была высказана более 100 лет назад, пока не имеет убедительной экспериментальной модели. Как видно из изложенного, в целом они могут дополнять друг друга. Патологические процессы в сосудах резистивного типа Как указывалось выше, задачей резистивных сосудов является поддержание на определенном уровне артериального давления. Отсюда следует, что патология резистивных сосудов проявляется прежде всего значительными отклонениями от нормы уровня артериального давления. Соответствующие патологические состояния называют гипер- и гипотензией. В первом случае уровни систолического и диастолического давления превышают соответственно 160 и 95 мм рт. ст. Во втором – уровень артериального давления меньше 100/60 мм рт. ст Уровень давления от 140/90 до 159/94 мм рт. ст. считается "опасной зоной" (пограничная гипертензия). Артериальная гипертензия – стойкое повышение артериального давления. По происхождению различают артериальную гипертензию первичную и вторичную. Вторичное повышение артериального давления является лишь симптомом (симптоматическая гипертензия), следствием какого-нибудь другого заболевания (гломерулонефрит, сужение дуги аорты, аденома гипофиза или коркового вещества надпочечных желез и т. д.). Первичную гипертензию до сих пор называют эссенциальной гипертензией, что указывает на невыясненность ее происхождения Гипертоническая болезнь является одним из вариантов первичной артериальной гипертензии. При первичной гипертензии повышение артериального давления является основным проявлением болезни. На долю первичной гипертензии приходится 80% всех случаев артериальной гипертензии. Остальные 20% составляют вторичную артериальную гипертензию, из них 14% связаны с заболеваниями паренхимы почек или ее сосудов. Экспериментальные модели. В эксперименте на животных стойкого повышения артериального давления можно добиться, последовательно влияя на различные звенья системы нейрогуморальной регуляции сосудистого тонуса. Выраженное и длительное повышение артериального давления развивается при одно- и двусторонней ишемии головного мозга, которая наступает после перевязки питающих мозг артерий (позвоночных, сонных артерий или ее ветвей). Это центрально-ишемическая гипертензия, обусловленная нарушением функционального состояния корковых и подкорковых центров регуляции сосудистого тонуса. Аналогичного эффекта можно добиться путем введения в мозжечково-мозговую (большую) цистерну мозга животных каолина, частицы которого блокируют пути оттока лимфы по периневральным и периваскулярным лимфатическим путям. С одной стороны, возникает повышение внутричерепного давления, с другой – некоторая степень ишемии. Вероятно, и то, и другое имеет значение в возникновении данного типа артериальной гипертензии. У высокоорганизованных животных (собаки, обезьяны) удалось получить гипертензию путем "сшибки", т. е. столкновения процессов торможения и возбуждения (воздействие дифференцировочного раздражителя вслед за положительным без обычного между ними интервала, столкновение пищевого и оборонительного рефлексов). Развивающаяся гипертензия является следствием невроза. Возникновение артериальной гипертензии возможно при психоэмоциональном стрессе, в условиях зоосоциального конфликта. Вторая группа моделей основана на повреждении депрессорных систем. Гейманс в 1931 – 1937 гг. и Н. Н. Горев в 1939 г. получилирефлексогенную гипертензию, или "гипертонию расторможения", после двусторонней перерезки у кроликов и собак депрессорных нервов и синусных ветвей языкоглоточного нерва. Механизм ее возникновения связан со снижением тормозящей ("сдерживающей") импульсации с рефлексогенных зон области дуги аорты и сонной пазухи. Разрушение ядра солитарного тракта с помощью электролитического воздействия дает такой же результат. К депрессорным факторам относятся простагландины A, Е1–2, I2. Естественно, что подавление их синтеза (индометацином) сопровождается повышением артериального давления. Почечные модели. В 1934 г. Гольдблатт воспроизвел хроническую гипертензию путем частичного сужения просвета обеих почечных артерий (реноваскулярная гипертензия). Эта модель гипертензии имеет ряд особенностей: во-первых, она удается лишь при частичном сужении просвета почечных артерий; во-вторых, ее можно воспроизвести лишь при ограничении поступления крови в обе почки. Одностороннее нарушение почечного кровообращения приводит, как правило, к преходящему повышению давления. Однако, если при этом удалить вторую (нормальную) почку, развивается стойкое повышение артериального давления. Наконец, длительную гипертензию можно получить удалением обеих почек (ренопривная гипертензия), переведя животных на гемодиализ или перитонеальный диализ для предотвращения уремии. Прессорным действием на сосуды обладают гормон мозгового вещества надпочечных желез – адреналин и вырабатываемый особыми нейронами гипоталамуса - вазопрессин. Если эти гормоны вводить в организм длительно, а главное, регулярно, у подопытных животных развивается гипертензия. Ее развитие связывают главным образом с прямым влиянием адреналина и вазопрессина на мышечные элементы артериальных сосудов. Кроме того, определенное значение при этом имеет стимуляция ими симпатической части вегетативной нервной системы с выделением норадреналина из окончаний симпатических нервов. В эксперименте на животных доказана также роль гормонов коркового вещества надпочечных желез в развитии артериальной гипертензии [Селье, 1943]. Особое значение при этом имеют минералокортикоиды – дезоксикортикостерони альдостерон. Хроническое введение их в умеренных дозах чувствительным животным (крысам, собакам, кроликам) с одновременным назначением им вместо питьевой воды раствора натрия хлорида приводит к значительному гипертензивному эффекту. Исключение натрия хлорида из воды и пищи приводит к тому, что артериальное давление в ответ на введение дезоксикортикостерона или альдостерона не повышается. Считают, что непосредственной причиной гипертензии является увеличение содержания натрия в стенке сосудов. Введение натрия хлорида не только способствует развитию минералокортикоидной гипертензии, но и в состоянии вызвать ее без каких-либо дополнительных воздействий (солевая гипертензия). Важно отметить, что у 2/3 животных (крыс) после отмены солевой диеты гипертензия оставалась. Существует прямая зависимость между уровнем артериального давления и суточной дозой натрия хлорида, длительностью его приема, возрастом животных (молодые животные более склонны к развитию солевой гипертензии) и наследственным предрасположением. Выведены линии крыс, предрасположенные или резистентные к развитию солевой гипертензии. У чувствительных линий животных имеются генетические дефекты или функции почек, или синтеза стероидов, что приводит к задержке натрия в организме. Другие варианты генетически обусловленной артериальной гипертензии основаны на предрасположенности определенных линий животных (крысы) к частым инсультам (до 80%), артериолипидозу сосудов мозга, на наличии наследственных дефектов синтеза простагландинов (дефицит простагландин-15-гидроксидегидрогеназы), на гиперчувствительности а-адренэргических структур гладких мышц сосудов к катехоламинам. Этиология. Причины первичной гипертензии, возможно, различны и многие из них до сих пор окончательно не установлены. Однако не подлежит сомнению, что определенное значение в возникновении гипертензии имеет,перенапряжение высшей нервной деятельности под влиянием эмоциональных воздействий. Об этом свидетельствуют частые случаи развития первичной гипертензии у людей, переживших ленинградскую блокаду, а также у людей "стрессовых" профессий. Особое значение при этом имеют отрицательные эмоции, в частности эмоции, не отреагированные в двигательном акте, когда вся сила их патогенного воздействия обрушивается на систему кровообращения. На этом основании Г. Ф. Ланг назвал гипертоническую болезнь "болезнью неотреагированных эмоций". Гипертоническая болезнь – это "болезнь осени жизни человека, которая лишает его возможности дожить до зимы" (А. А. Богомолец). Тем самым подчеркивается роль возрастав происхождении гипертонической болезни. Однако и в молодом возрасте первичная гипертензия встречается не так редко. Важно при этом отметить, что до 40 лет мужчины болеют чаще, чем женщины, а после 40 соотношение приобретает противоположный характер. Определенную роль в возникновении первичной гипертензии играет наследственный фактор. В отдельных семьях заболевание встречается в несколько раз чаще, чем у остального населения. О влиянии генетических факторов свидетельствует и большая конкордантность по гипертонической болезни у однояйцевых близнецов, а также существование линий крыс, предрасположенных или резистентных к некоторым формам гипертензии. В последнее время в связи с проведенными в некоторых странах и среди народностей (Япония, Китай, негритянское население Багамских островов, некоторые районы Закарпатской области) эпидемиологическими наблюдениями установлена тесная связь между уровнем артериального давления и количеством потребляемой соли. Считают, что длительное потребление более 5 г соли в день способствует развитию первичной Гипертензии у людей, имеющих наследственное предрасположение к ней. Успешное экспериментальное моделирование "солевой гипертензии" подтверждает значение избыточного потребления соли. С приведенными наблюдениями хорошо согласуются клинические данные о благоприятном терапевтическом эффекте низкосолевой диеты при некоторых формах первичной гипертензии. Таким образом, в настоящее время установлено несколько этиологических факторов гипертензии. Неясно только, какой из них является причиной, а какой играет роль условия в возникновении болезни. Патогенез. Согласно уравнению Пуазейля Р = Q•R, давление жидкости в системе трубок (Р) определяется величиной их наполнения (Q) и сопротивления току жидкости (R). Применительно к системе артериальных сосудов это должно означать, что давление в них определяется в основном двумя факторами: величиной минутного объема крови сердца (количеством крови, выбрасываемым левым желудочком за 1 мин – (Q)) и сопротивлением, которое кровоток встречает в сосудах (R). В свою очередь величина минутного объема крови сердца (Q) определяется массой циркулирующей крови (в норме 4 – 5 л), систолическим выбросом сердца (в норме 70 мл), частотой сердечных сокращений (в норме 70/мин), венозным возвратом крови к сердцу, а периферическое сопротивление кровотоку (R) зависит от диаметра сосудов (пассивные и активные изменения), вязкости крови, ее трения о стенки, наличия вихревых движений. В зависимости от того, увеличение какого из приведенных двух параметров (Q или R) определяет повышение артериального давления, различают следующие гемодинамические варианты артериальной гипертензии. 1. Гиперкинетический тип – увеличенный минутный объем крови сердца (Q), неизмененное или слегка пониженное периферическое сопротивление кровотоку (R). 2. Эукинетический тип – связан с увеличением как Q, так и R. 3. Гипокинетический тип – величина Q не изменена или несколько уменьшена, зато резко увеличено R.. В сущности гипокинетический тип артериальной гипертензии соответствует понятию "гипертоническая болезнь" (Г. Ф. Ланг, А. Л. Мясников). Что касается других гемодинамических вариантов артериальной гипертензии, то полагают, что увеличение сердечного выброса имеет место на ранних стадиях заболевания, после чего гиперкинетический вариант переходит в эукинетический и гипокинетический. Основные механизмы, определяющие становление гиперкинетического типа (или фазы) повышения артериального давления, сводятся или к усилению положительного хроно- и инотропного эффекта вследствие усиления нейрогуморальных влияний на сердце (активация ?-адренорецепторов), или к повышению венозного возврата крови к сердцу из-за веноконстрикции (?-адренорецепторный механизм) с перераспределением кровотока из периферии в крупные венозные коллекторы. Таким образом, представляется, что стресс, как один из основных этиологических факторов первичной артериальной гипертензии, вызывает активацию гипофизарно-надпочечниковой и симпатоадреналовой системы с последующим включением ренин-ангиотензин-альдостеронового прессорного механизма. Это приводит к увеличению минутного объема крови сердца при неизменной или слегка сниженной величине общего периферического сопротивления. На втором этапе патогенеза увеличение кровяного давления и местного кровотока приводит согласнозакону Остроумова – Бейлиса к увеличению сосудистого тонуса и генерализованному спазму артериол, направленному на приведение в соответствие местного кровотока и потребности в нем тканей. В последующем длительное повышение сосудистого тонуса приводит к гипертрофии сосудистых гладких мышц, что в еще большей степени увеличивает общее периферическое сопротивление сосудов и выраженность артериальной гипертензии. Этому способствует и повышение содержания Na+и воды в гладких мышцах, повышение их чувствительности в этих условиях к различным прессорным агентам. Следует думать, что первичная (эссенциальная) артериальная гипертензия ни по этиологии, ни по патогенезу не является единым заболеванием. В каждом конкретном случае ее характер определяется характером и степенью нарушения прессорных и депрессорных механизмов, участием нервного, почечного и гормонального факторов в возникновении подобных нарушений. Нервный фактор. В наиболее последовательной форме роль нейрогенного фактора в патогенезе гипертонической болезни представлена в концепции Г. Ф. Ланга и А. Л. Мясникова. Они считали, что "нервное перенапряжение, которое составляет этиологическую сущность гипертонической болезни, реализуется именно в расстройстве трофики определенных мозговых центров управления артериальным давлением" [Мясников А. Л., 1965], прежде всего тех структурных образований коры большого мозга (передний полюс лобной доли, глазничные извилины, двигательная зона коры, передний полюс височной доли, крестовидная извилина гиппокампа, миндалевидное тело), раздражение которых, как это показали эксперименты на животных, вызывает повышение артериального давления. Нарушение корковой динамики, изменение соотношения между процессами возбуждения и торможения структур коры большого мозга приводят к истощению их функций, к развитию, наряду с различного рода невротическими расстройствами и нарушениями высшей нервной деятельности, длительного патологического повышения артериального давления. Непосредственный механизм повышения артериального давления связан с "буйством" подкорки, устойчивым возбуждением вегетативных центров гипоталамуса, в первую очередь сосудодвигательного центра. Различные дополнительные влияния усиливают очаг патологической доминанты в области сосудодвигательного центра, тормозя по принципу отрицательной индукции функции других центров. Вазомоторные импульсы, зарождающиеся в гипоталамусе – высшем интеграторе вегетативных функций, достигают ядер продолговатого мозга, затем по симпатическим нервным путям – сосудов, вызывая усиление вазомоторного компонента сосудистого тонуса. Это осуществляется посредством норадреналина – нервного медиатора, который, активируя а-адренэргические рецепторы артериол, повышает их сопротивление кровотоку, не оказывая существенного прямого влияния на миокард. Частично гипертензивный эффект может быть обусловлен и возбуждением мозгового вещества надпочечных желез под влиянием симпатической части вегетативной системы с поступлением в кровь избытка адреналина инорадреналина. Для развития гипертензии в последнем случае необходимо, однако, чтобы ?-адренэргическое действие адреналина и норадреналина суммарно превышало ?-адренэргическое действие адреналина, направленное на снижение тонуса мышечных структур артериол. Иными словами, сосудосуживающий эффект норадреналина, связанный с возбуждением ?-адренорецепторов резистивных сосудов, и увеличение сердечного выброса, обусловленное возбуждением ?-адренорецепторов сердца под действием адреналина, в сумме должны перекрывать ?-адренэргическое сосудорасширяющее действие последнего. Клинический опыт, однако, показал, что подобная ситуация складывается лишь в 15 – 25% случаев гипертонической болезни. У остальных больных отсутствует прямые признаки усиления активности симпатоадреналовой системы, что заставляет предположить существование других механизмов повышения сосудистого тонуса. В частности, допускают, что в результате длительных центрогенных спазмов артериол в дальнейшем развиваются вторичные патологические изменения сосудов (в основном почечных), которые включают в патогенез ренин-ангиотензиновую систему по принципу порочного круга. В тесной связи с деятельностью центральных механизмов регуляции сосудистого тонуса находится функционирование периферических барорецепторов сонной пазухи и дуги аорты. Их выключение, как это вытекает из опытов Гейманса, сопровождается выраженной и длительной прессорной реакцией. С этим фактом хорошо согласуется наблюдение П. К. Анохина, сущность которого заключается в том, что при гипертензии (например, почечной) с течением времени снижается чувствительность барорецепторов сонной пазухи. Кроме того, установлено, что устранение тормозящего влияния барорецепторов на сосудодвигательный центр, а также на сетчатое образование мозгового ствола и гипоталамуса вызывает четырехкратное увеличение секреции катехоламинов мозговым веществом надпочечных желез и значительное усиление секреции альдостерона корковым веществом. Последнее свидетельствует о принципиальной возможности изменений и миогенного компонента сосудистого тонуса при гипертензии нейрогенного происхождения. Однако они, как и изменения со стороны почек, имеют скорее вторичный генез, так как всегда следуют за повышением артериального давления, а не наоборот. Почечный фактор. Как было отмечено выше, опыты Гольдблатта показали, что нарушение почечного кровообращения сопровождается артериальной гипертензией. Доказано также, что при гипертонической болезни у человека изменения со стороны почек также могут играть определенную роль в прогрессировании заболевания. В настоящее время известно, что почка может способствовать как повышению, так и понижению артериального давления. Первое наступает при стимуляции функции юкстагломерулярного (околоклубочкового) аппарата почек. В его клетках при этом образуется ренин-протеолитический фермент, субстратом для которого служит ?2-глобулин плазмы крови – ангиотензиноген. Секреция ренина зависит от степени растяжения приносящих сосудов клубочков. Понятно, что нарушение кровообращения в почках (ишемия, снижение пульсового и среднего артериального давления) стимулирует этот процесс. Увеличение выработки ренина сопровождается увеличением в крови концентрации ангиотензина I – продукта ферментативной реакции ангиотензиноген – ренин. Этот полипептид (декапептид) под действием конвертирующего фермента крови превращается в ангиотензин II(октапептид). Последний в отличие от ангиотензина I обладает прямым влиянием на стенку преимущественно прекапиллярных сосудов. Частично сосудосуживающий эффект ангиотензина II опосредуется также симпатической частью вегетативной нервной системы (усиление функции), корковым веществом надпочечных желез (увеличение секреции альдостерона) и почек (усиление реабсорбции ионов натрия). Это вещество является наиболее мощным из всех известных в настоящее время прессорных агентов (рис. 19.14). 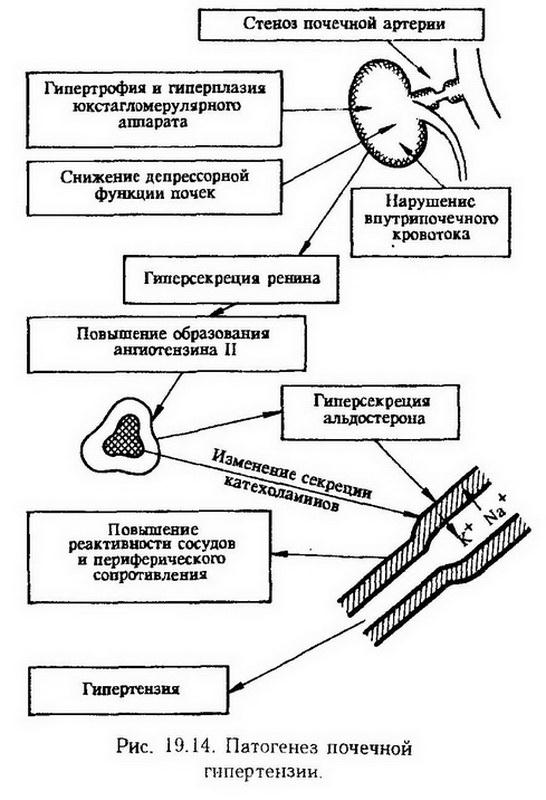 В крови и тканях (главным образом в почках) под действием фермента ангиотензиназы ангиотензин II быстро разрушается. Здоровая почка (при нарушении кровообращения в другой почке) образует повышенное количество ангиотензиназы, тем самым предотвращая развитие стойкой гипертензии. При нарушении гемодинамики в обеих почках их ангиотензиназная активность резко снижается, что способствует стойкому повышению артериального давления. Следовательно, при нарушении кровообращения в почках артериальная гипертензия частично обусловлена ренин-ангиотензиновой системой, частично – снижением выработки специфических и неспецифических ангиотензиназ. Однако повышенная секреция ренина ишемизированными почками и повышенное количество ангиотензина в крови наблюдается лишь в первые дни после сужения почечных артерий, в то время как гипертензия удерживается на высоком уровне в течение длительного времени. Этот факт, а также появление стойкой гипертензии после удаления обеих почек привели к предположению (Гроллмен), что хроническая гипертензия может быть обусловлена отсутствием (нарушением) какой-то депрессорной функции почек (ренопривная гипертензия) или (и) действием каких-то внепочечных веществ. Это предположение полностью подтвердилось. В настоящее время считается общепризнанным, что нормальные почки обладают способностью контролировать действие как почечных, так и экстраренальных прессорных веществ (вазопрессин, катехоламины, альдостерон), т. е. антипрессорной (противогипертензивной) функцией. Считают, что эта функция осуществляется с помощью веществ липидной природы: фосфолипидного ингибитора ренина, нейтральных жиров из мозгового вещества почек и простагландинов типа А и Е. Следовательно, выключение этой функции почек также ведет к преобладанию вазопрессорных механизмов и, таким образом, к гипертензии. Особый интерес представляют простагландины – производные полиненасыщенных жирных кислот. Местом их образования в почке, по-видимому, служат интерстициальные звездчатые клетки мозгового вещества. Контроль за секрецией простагландинов осуществляется с помощью ангиотензина II. Повышенное количество его в плазме крови стимулирует образование простагландинов. Простагландины типа Е вызывают гипотензивный эффект при нормальном уровне артериального давления. Простагландины типа А не вызывают понижения давления у здоровых животных или людей, но предотвращают развитие почечной гипертензии Гольдблатта и ренопривной гипертензии у животных или снижают повышенное артериальное давление у человека (рис. 19.15). Помимо расширяющего действия на артериолы, простагландины вызывают усиленное выделение натрия с мочой, что также предотвращает развитие гипертензии. Аналогичным действием обладает и почечная калликреин-кининовая система. Приведенные данные убедительно свидетельствуют о важной патогенетической роли почечного фактора в развитии и стабилизации гипертензии. Следует, однако, отметить, что первичная гипертензия с точки зрения участия почечного прессорного фактора не является однородным понятием. В зависимости от уровня ренина в плазме крови различают по крайней мере три вида гипертензии: гиперренинемическую – на ее долю приходится 25 – 30% случаев гипертонической болезни, норморенинемическую – 55 – 60% и гипоренинемическую – 10 – 20% случаев. Гормональный фактор. Эндокринные железы участвуют в регуляции артериального давления в норме, нарушение их функции является важным звеном патогенеза гипертензии. Наибольшее значение в этом плане имеют надпочечные железы. Как уже было сказано, в эксперименте гипертензию можно получить введением альдостерона и дезоксикортикостерона, особенно сочетая с нагрузкой натрия хлоридом. Альдостероновый механизм принимает участие в поддержании повышенного артериального давления и у человека. Считают, что основные изменения в организме, вызываемые дезоксикортикостероном и альдостероном, связаны с их влиянием на обмен натрия. Под действием этих гормонов происходит задержка ионов натрия в организме в результате его усиленной реабсорбции из первичной мочи на всем протяжении канальцев, но особенно в их дистальной части. Следствием задержки солей натрия в организме является их накопление в мышечных элементах артерий. При этом в связи с одновременной задержкой воды развивается гиперволемия, а также отек эндотелия и частичное сужение просвета сосудов. Кроме того, в результате повышенной концентрации ионов натрия и кальция в сосудистой стенке увеличивается ее чувствительность к различным прессорным влияниям – нервным и гуморальным (катехоламины, вазопрессин, ангиотензин II). В физиологических условиях антагонистом альдостерона и ангиотензина II являетсянатрийуретический предсердный гормон. Это полипептид, состоящий из 28 аминокислот, синтезирующийся преимущественно в предсердиях, в меньших количествах – в желудочках сердца. Его антигипертензивное действие обусловлено прежде всего способностью усиливать натрийурез и диурез (угнетается реабсорбция натрия и воды в канальцах почек, усиливается клубочковая фильтрация). Кроме того, он угнетает выработку альдостерона и, наконец, обеспечивает выраженный сосудорасширяющий эффект за счет торможения входа кальция в гладкомышечные клетки и выброса Ca2+ из внутриклеточных депо. Следовательно, высокий уровень как миогенного компонента сосудистого тонуса, так и вазомоторного достигается в условиях, когда баланс альдостерона и ангиотензина II, с одной стороны, и натрийуретического предсердного гормона, с другой, смещается в пользу первых. Такой дисбаланс возникает уже на ранних стадиях артериальной гипертензии и является важным фактором ее стабилизации. Стойкая гипертензия некоторое время может не сопровождаться выраженной клинической симптоматикой. Клинический опыт, однако, показывает, что при длительном течении гипертензия осложняется первоначально недостаточностью местного кровообращения, а в дальнейшем и недостаточностью соответствующих органов и даже систем. Наиболее частыми клиническими формами осложненной первичной гипертензии являются недостаточность мозгового кровообращения, включая инсульт, гипертрофия миокарда с последующей недостаточностью кровообращения, недостаточность почечного кровообращения и почечная недостаточность (синдром первично сморщенной почки). Легочная гипертензия характеризуется повышением легочного артериального давления сверх 25 мм рт. ст., служит причиной развития так называемого легочного сердца (cor pulmonale) и недостаточности кровообращения по правожелудочковому типу. По своему происхождению может быть первичной и вторичной. Возникновение вторичной легочной гипертензии связывают с заболеваниями, первично поражающими воздухоносные пути и альвеолы (хронический бронхит, бронхиальная астма, хроническая пневмония, эмфизема легких, фиброз легких), первично нарушающими подвижность грудной клетки (кифосколиоз, фиброз плевры, хроническая нервно-мышечная слабость, например, при миастении, полиомиелите), а также заболеваниями, первично поражающими легочные сосуды (узелковый периартериит, тромбоз мелких кровеносных сосудов, эмболия легочной артерии и др.) и сердце (митральный стеноз, дефект межжелудочковой перегородки). Более редко встречающаяся первичная легочная гипертензия не имеет каких-либо известных причин, развивается при отсутствии заболеваний легких и сердца (легочная гипертензия неизвестной этиологии). В некоторых случаях первичная легочная гипертензия является врожденной. Ведущим механизмом развития первичной легочной гипертензии является снижение рO2 в альвеолярном воздухе. В высокогорных условиях влияние этого фактора, естественно, выражено сильнее. Низкое парциальное давление кислорода в альвеолярном воздухе оказывает непосредственное влияние на гладкомышечные элементы легочных сосудов (артерий), обусловливая стойкое повышение их тонуса. Один из возможных механизмов такого прямого влияния – потеря кальция и поглощение натрия сосудистой стенкой, деполяризация мембран и снижение порога возбудимости. Вазопрессорный эффект гипоксии усиливается при развитии ацидоза и физической нагрузке. С другой стороны, водородным ионам, вазоактивным аминам (гистамин, серотонин), метаболитам, образующимся при гипоксии непосредственно в мышечных клетках легочных сосудов, а также поступающим в них из периферических органов и тканей, приписывают роль посредников прессорного эффекта гипоксии на легочные сосуды [Хомазюк А. И., 1978]. Алкалоз ослабляет вазопрессорный эффект гипоксии. Сосудорасширяющее действие на легочные артерии оказывают аденозин, АМФ, ацетилхолин. Действие брадикинина вариабельно. Полагают, что нейрогенные механизмы играют меньшую роль в развитии первичной легочной гипертензии в ответ на гипоксию или возникающей по какой-либо другой (неизвестной) причине. Это обусловлено тем, что к мелким легочным артериям мышечного типа подходит мало нервов (или их нет вообще), кроме того, нервы, как правило, оканчиваются не в медии, а в адвентиции. Раздражение нервов сопровождается повышением упругости стенок крупных легочных артерий, но не повышением сопротивления кровотока в области легочных артерий мышечного типа. Сужение легочных сосудов при гипоксии имеет место и в условиях денервации легких. Дискутируется вопрос относительно возможности развития первичной легочной гипертензии у отдельных лиц в связи с развитием тромбоэмболического синдрома, нарушением способности легких поглощать вазоактивные вещества (простагландины, серотонин, гистамин, норадреналин и др.), а также в связи с индивидуальной повышенной реактивностью сократительных элементов легочных сосудов на гипоксию или другие вазоактивные факторы. Артериальная гипотензия. В отличие от гипертензии артериальная гипотензия представляет собой стойкое понижение артериального давления, обусловленное преимущественно понижением тонуса резистивных сосудов. Наблюдается она чаще у лиц астенической конституции, характеризуется понижением физического развития и питания, общей адинамией, быстрой утомляемостью, тахикардией, одышкой, головокружением, головной болью, обмороками. В настоящее время выделяют артериальную гипотензию физиологическую (не сопровождается болезненными симптомами) и патологическую (с характерным симптомокомплексом). Последняя бывает острой и хронической. Хроническая артериальная гипотензия подразделяется на симптоматическую (вторичную) и нейроциркуляторную дистонию гипотензивного типа (первичную гипотензию). Патогенетически, учитывая, что уровень артериального давления определяется величиной сердечного выброса, количеством циркулирующей крови и тонусом резистивных сосудов, возможны три гемодинамические формы артериальной гипотензии: связанная с недостаточностью сократительной функции сердца; вызванная уменьшением количества циркулирующей крови и возникающая вследствие понижения тонуса резистивных сосудов. Симптоматическая хроническая артериальная гипотензия (вторичная) является следствием ряда общих соматических острых и хронических заболеваний: сердца (пороки, миокардит, инфаркт миокарда); головного мозга (комоция), легких (крупозная пневмония), печени (гепатит, механическая желтуха), крови (анемия), эндокринных желез, а также экзогенных и эндогенных интоксикаций. Что касается происхождения нейроциркуляторной (первичной) артериальной гипотензии, то по аналогии с гипертонической болезнью считают, что основным этиологическим и патогенетическим фактором первичной артериальной гипотензии также является перенапряжение основных процессов коры большого мозга (возбуждения и торможения). Однако в отличие от первичной гипертензии наблюдается превалирование торможения и распространение его на подкорковые вегетативные образования, в частности на сосудодвигательный центр. Ослабление вследствие этого эффективных сосудосуживающих влияний на фоне свойственного астеническому типу конституции преобладания холинэргических влияний над адренэргическими является непосредственной причиной снижения тонуса резистивных сосудов, периферического сопротивления и артериального давления. Патологические изменения в сосудах обмена рассмотрены в разделе "Нарушения микроциркуляции". Шок Шок (от англ, shock – удар, сотрясение) представляет собой тяжелый патологический процесс, сопровождающийся истощением жизненно важных функций организма и приводящий его на грань жизни и смерти вследствие критического уменьшения капиллярного кровообращения в пораженных органах. В соответствии с современными представлениями об основных этиологических факторах и механизмах шока выделяют следующие его формы: 1. Первичный гиповолемический шок. Возникновение гиповолемического шока связано с наружной или внутренней кровопотерей (травма, в том числе операционная, повреждение органов и тканей патологическим процессом, нарушение свертывания крови); потерей плазмы (ожоги, размозжение тканей); потерей жидкости и электролитов (кишечная непроходимость, панкреатит, перитонит, энтероколит, перегревание); перераспределением крови в сосудистом русле (тромбоз и эмболия магистральных вен). Остро возникающий дефицит объема крови при этом приводит к уменьшению величины венозного возврата к сердцу, к снижению ударного и минутного объема крови сердца (УО, МО) и артериального давления. За счет симпатоадренэргической реакции (стимуляция р-рецепторов сердца и а-рецепторов периферических кровеносных сосудов) обеспечивается увеличение частоты сердечных сокращений и повышение периферического сопротивления сосудов с целью нормализации артериального давления и кровоснабжения, прежде всего сердца и головного мозга. Недостаточность указанных механизмов, как и отрицательные последствия вазоконстрикции, сопровождаются резким уменьшением кровоснабжения органов и тканей и характерными проявлениями шока. 2. Травматический шок. Возникновение и течение травматического шока характеризуют некоторые существенные особенности. Так, травматический шок развивается на фоне резко выраженного раздражения и даже повреждения экстеро-, интеро- и проприорецепторов вследствие прямого повреждающего действия физических факторов и существенных нарушений функций центральной нервной системы. Эти нарушения характеризуются стадийностью течения (стадия возбуждения, или эректильная – от лат. erectus – напряженный, стадия торможения, или торпидная, – от лат. torpidus – оцепенелый). Стадия возбуждения кратковременна, ее отличает состояние возбуждения центральной нервной системы (кора, подкорковые образования, вегетативные ядра симпатической нервной системы), следствием которого является усиление функции системы кровообращения, дыхания, некоторых эндокринных желез (гипофиз, мозговое и корковое вещество надпочечных желез, нейросекреторные ядра гипоталамуса) с высвобождением в кровь избыточного количества кортикотропина, адреналина, норадреналина, вазопрессина и развитием стрессового синдрома. Стадия торможения более продолжительна (от нескольких часов до суток) и характеризуется развитием в центральной нервной системе тормозных процессов, уравнительной и парадоксальной стадий парабиоза с распространением указанных процессов на отделы мозгового ствола, гипоталамус и спинной мозг и снижением функций жизненно важных органов и систем. Указанные представления легли в основу нервно-рефлекторной теории патогенеза травматического шока. В механизме возникновения и развития травматического шока определенную роль играет токсемия, обусловленная всасыванием в систему кровообращения продуктов распада и лизиса нежизнеспособных тканей. Доказательства значения этого фактора получены В. Кенноном на примере"турникетного" шока, возникающего после снятия жгута спустя четыре и более часов после его наложения или после прекращения длительного сдавливания частей тела (при обвалах шахт, рудников, больших земляных массивов, при землетрясении, бомбардировке и т. д.). В последние годы из поврежденных тканей и плазмы крови выделена большая группа тканевых олигопептидов, ответственных за развитие связанных с токсемией патофизиологических сдвигов. Указанные олигопептиды, однако, неспецифичны. Травматический шок не всегда сопровождается абсолютной потерей крови или плазмы. На этом основании такую разновидность травматического шока долгое время ошибочно считали изоволемическим шоком. При этом не учитывалась возможность дисволемических изменений, обусловленных понижением эффективности циркулирующего объема крови вследствие ее застоя в определенных сосудистых областях или повышенной транссудацией жидкой части крови. 3. Кардиогенный шок наблюдается при снижении насосной функции сердечной мышцы (инфаркт миокарда, миокардит), при тяжелых нарушениях сердечного ритма (пароксизмальная тахикардия, синдром Морганьи – Адамса – Стокса), при тампонаде сердца (тромбоз полостей, выпот или кровотечение в околосердечную сумку), при массивной эмболии легочной артерии (тромбоэмболия легких, жировая эмболия). Ведущим механизмом кардиогенного шока является уменьшение производительности сердца в связи с миогенным нарушением его насосной функции или с наличием препятствий для заполнения желудочков. Следствием этого является уменьшение ударного и минутного объема крови, артериального давления, с одной стороны, и увеличение давления наполнения сердца, с другой. Как и при гиповолемическом шоке, вследствие симпатоадренэргической реакции наблюдается тахикардия и увеличение периферического сопротивления сосудов, которые лишь усугубляют гемодинамические нарушения из-за недостаточной насосной функции сердца. 4. Сосудистые формы шока. К ним относится септический и анафилактический шок. Септический, или инфекционно-токсический, шок возникает при инфекциях, вызванных чаще всего грамотрицательной (кишечная палочка, протей), реже грамположительной микробной флорой (стафилококк, стрептококк). Анафилактический шок развивается вследствие повышенной чувствительности организма к веществам антигенной природы. Общим механизмом в развитии сосудистых форм шока является первичное нарушение сосудистой регуляции, которое, однако, совершенно различно при обеих его формах. Так, при септическом шоке вследствие действия бактериальных токсинов первичные расстройства периферического кровообращения развиваются в связи с открытием артериовенозных шунтов. При этом кровь устремляется из артериального русла в венозное в обход капиллярной сети. Нарушение питания тканей, вызванное ограниченностью капиллярного кровотока, усугубляется прямым влиянием бактериальных токсинов на метаболизм тканей, в частности на потребление кислорода. Общее периферическое сопротивление и артериальное давление при септическом шоке в связи с открытием артериовенозных шунтов резко понижено, давление наполнения сердца нормально или повышено. Компенсаторно, особенно в начальной фазе шока, увеличивается ударный объем крови и частота сердечных сокращений, вследствие чего увеличивается минутный объем крови. Однако в связи с развитием миокардиальной формы недостаточности сердца и нарастающим дефицитом циркулирующего объема крови в поздних фазах септического шока основные показатели деятельности сердца (УО, МО) также резко снижаются. При анафилактическом шоке в связи с накоплением гистамина и других вазоактивных веществ (кинины, серотонин и др.) происходит резкое уменьшение сосудистого тонуса, снижение артериаль ного давления. Снижается давление наполнения сердца из-за уменьшения венозного возврата крови к сердцу. Причиной этого является расширение капиллярных и емкостных сосудов венозного отдела кровеносного русла. Скопление крови в капиллярных сосудах и венах приводит к уменьшению объема циркулирующей крови и к относительной гиповолемии. Наблюдается и прямое нарушение сократительной деятельности сердца. Симпатоадренэргическая реакция при этом не выражена вследствие нарушений сосудистого тонуса. Все вместе определяет катастрофический характер течения анафилактического шока. Существенные особенности патогенеза анафилактического шока связаны с видовой принадлежностью организма (см. раздел VII "Аллергия"). Следствием макрогемодинамических нарушений независимо от разновидности шока и от той последовательности, в которой они возникают, являются нарушение микроциркуляции, в частности уменьшение капиллярного кровотока, нарушение доставки к тканям кислорода, энергетических субстратов, затруднение выведения конечных продуктов обмена веществ. Развивающийся при этом метаболический ацидозвызывает дальнейшие расстройства микроциркуляции вплоть до полной остановки движения крови. С ним связаны, в частности, расширение прекапиллярных артериол, увеличение экссудации жидкости из крови в ткани, набухание и агрегация клеток крови, повышение вязкости крови, увеличение свертываемости крови и диссеминированное микротромбообразование в капиллярных сосудах. Тотальное нарушение функций клеток, прежде всего деятельности натрий-калиевого насоса, биосинтетической активности, целости лизосомального аппарата, ставящие организм на грань жизни и смерти, является конечным результатом описанных выше нарушений микроциркуляции при сосудистых формах шока. Особенно чувствительны к расстройствам микроциркуляции при шоке легкие, почки, печень. В связи с этим нередким осложнением шока является острая недостаточность дыхания, почек или печени. Предшествующие заболевания (лучевая болезнь, анемия, голодание и др.) снижают толерантность организма к шоку. Понижена она также в детском возрасте в связи с особенностями физиологического развития детского организма. Коллапс В узком смысле понятие "коллапс" используется для обозначения резкого падения артериального давления (ниже некоторого критического значения – тем большего, чем выше исходный тонус), вследствие чего сосуды утрачивают устойчивость формы, спадаются ("схлопываются"). Клинически коллапс характеризуется кратковременной потерей сознания, общим упадком сил, патофизиологически – признаками острой сосудистой недостаточности кровообращения с нарушениями гемодинамики практически во всех органах и тканях и угнетением жизненных функций. В основе его развития лежит несоответствие между объемом циркулирующей жидкости и величиной просвета сосудов. Таким образом, причинами развития коллапса могут быть как внезапное уменьшение объема крови (кровопотеря, обезвоживание), так и внезапное расширение сосудистого ложа – вазомоторный коллапс (при повышении тонуса блуждающего нерва, при ортостатической дисрегуляции, при истощении ос-адренореактивности резистентных сосудов). Уже этой краткой характеристики достаточно, чтобы заключить, сколь трудно дифференцировать шок и коллапс. Здесь ориентиром должны служить наличие и выраженность дисметаболических клеточных и тканевых нарушений. При шоке они налицо и их тяжесть в динамике шока закономерно и прогрессивно нарастает. При коллапсе превалируют гемодинамические расстройства, однако они являются кратковременными и исчезают спонтанно, хотя в части случаев могут напоминать и картину шока. |